|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
en travaux décembre 2009 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Plan |
|
|
|
Sources générales:
Web: Un incontournable: le "moteur de
recherche sur les sciences du vivant"
(en anglais)
qui fournit essentiellement
des données moléculaires, y compris
phylogénétiques
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
2 sens ... :
|
|
ensemble des gènes1 d'un organisme |
|
ensemble du matériel génétique d'un organisme |
|
Au sens strict on ne devrait parler que de génome d'un être vivant, mais, par extension, on parle du "génome" d'un organite (« génome mitochondrial ») ou d'un virus (« génome viral ») alors qu'il s'agit clairement des acides nucléiques (ADN et ARN) sans que l'on puisse parler de matériel "génétique". |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
1un gène est une unité fonctionnelle (synthétique) de l'ADN qui correspond à un produit (ARN ± protéine). |
|
Par « matériel génétique » on entend « acides nucléiques (ADN et ARN) et produits associés directement » (comme les protéines se fixant sur les séquences régulatrices ou encore les histones structurant l'ADN eucaryote...). C'est une définition "technique" (chimique) d'une grande utilité pratique. Elle a des implications théoriques lorsque l'on s'efforce de comprendre la fonction de grandes portions du génome, sans rechercher les gènes. |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
... correspondant plus ou moins à deux approches de la génomique - science des génomes - : |
|
La génomique fonctionnelle en cherchant les fonctions des génomes peut s'intéresser au gènes mais aussi au transcriptome et au protéome. Elle ne se différencie guère alors de la génétique fonctionnelle excepté par le fait qu'elle travaille sur de plus grandes portions d'ADN... |
|
La génomique structurale cherche à connaître l'organisation du génome chez un organisme (localisation, disposition, éléments associés, sous-parties...). |
|
Mais la génomique peut aussi aller plus loin que ce clivage et ne pas se contenter des outils et du niveau de compréhension de la génétique. L'approche mathématique fonctionnelle rejoint alors l'approche structurale (morphologique) "à la René Thom". C'est une des voies nouvelles à explorer. On a dans ce cas deux niveaux possibles: les molécules (chromatine par exemple) et les organites (comme les chromosomes). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
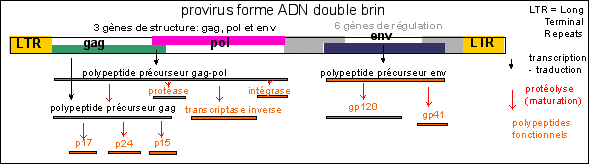
Des systèmes génétiques variés pour des rôles variés Un exemple
de virus (le VIH)
est étudié rapidement en terminale S à
l'occasion du
cours sur
l'immunité
|
|
Il est hors de question de présenter ici, de façon un tant soit peu complète, l'extrême diversité du matériel génétique des virus. Ce qui importe est bien leur signification biologique. Les virus sont des assemblages nucléoprotéiques organisés. Ce ne sont pas des êtres vivants (ne possèdent pas d'ADN et d'ARN simultanément, n'ont aucun métabolisme et, enfin, ne peuvent se multiplier sans cellules vivantes). Leur taille se situe approximativement entre celles d'un ribosome, pour les plus petits et d'une bactérie comme E. coli pour les plus grands. |
|
Lors de la phase extracellulaire, la particule virale complète est qualifiée de virion. Les formes de l'enveloppe sont souvent régulières et permettent d'élaborer une classification. Lors de la phase intracellulaire, seul l'ADN ou l'ARN viral pénétrant la cellule (provirus), il devient à la fois très simple, mais aussi très inexact de tenter de classer les virus à l'aide de leur « génome ». |
|
Du fait de leur extrême
variété et de leur grande similitude avec du
matériel génétique d'une cellule
hôte, comment savoir si un ADN ou un ARN trouvé
dans une cellule est bien vraiment étranger et
provient d'une particule virale ? Aussi, la réponse -
évidente -, qui met en avant la séquence,
est-elle très incomplète. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Des fragments de matériel génétique.... mais pas des génomes |
|
Certains petits ARN circulaires simple brin (de taille comprise entre 0,25 et 0,37kb; 1kb = 1.000 bases azotées, c'est-à-dire 1.000 nucléotides) trouvés dans le noyau de certaines cellules de plantes en un grand nombre d'exemplaires (200 à 10.000) sont qualifiés de viroïdes. Ils semblent responsables d'une dizaine de maladies dont par exemple, la maladie des tubercules en fuseau de la pomme de terre. On trouve ensuite des provirus, de
plus grande taille : |
|
Tous les types de provirus existent : ADN double brin, ADN simple brin, ARN simple brin, ARN double brin. Certains provirus sont linéaires, d'autres circulaires, certains sont fragmentés en plusieurs sous-unités (identiques ou non), d'autres présentent une alternance de zones simple- et double-brins, certaines extrémités des brins doubles pouvant se réunir (pontage). Enfin, certains ARN sont des chaînes positives (identiques à un ARNm viral et qui pourraient être traduites directement en protéines, mais qui cependant peuvent être d'abord rétrotranscrites en ADN), d'autres sont des chaînes négatives (complémentaires de l'ARNm viral). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
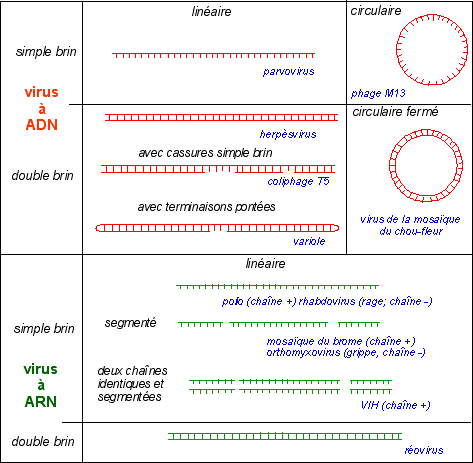 Exemples de types d'acides nucléiques viraux (d'après Microbiologie, Prescott, Harley, Klein, DeBoeck Université, 1995, Tab.17.1) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Pas de virus sans cellules... les virus seraient-ils des signaux intercellulaires ? |
|
Il n'est pas rare que l'on se contente du raisonnement analogique qui associe virus et êtres vivants : puisque le génome de ces derniers "code" pour les protéines, les acides nucléiques viraux codent probablement aussi pour les éléments de la capside protéique ou au moins sont nécessaire à son assemblage. Or, on est loin du compte. Certains éléments sont apportés par la cellule hôte sans que l'on puisse identifier un signal de commande venant du virus. Certaines séquences virales donnent des produits dont on ne connaît pas le rôle et l'auto-assemblage entre acides nucléiques et protéines de la capside- mécanisme qui semble être la règle - n'est plus ou moins compris que pour quelques virus. |
|
Il existe une autre
thèse, plus difficilement acceptable, et qui pourtant
reste fortement présente dans l'imaginaire de
beaucoup, selon laquelle les virus auraient
évolué à partir des procaryotes
(évolution rétrograde). Ils seraient en
quelque sorte d'anciens parasites devenus très
réduits. Les données actuelles nous
écartent franchement de cette hypothèse,
même si, comme dans toute théorie
évolutive, il ne peut y avoir de preuve
définitive (le passé est inaccessible à
l'expérience).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Quelques exemples...
|
|
Toutes les données sur les
génomes sont maintenant informatisées et
internationalisées sur le consortium pour le
génome : |
|
- la page NCBI sur les virus est à
l'adresse: http://www.ncbi.nlm.nih.gov/
genomes/ GenomesHome.cgi?
taxid=10239 Remarque: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
Certains virus peuvent contenir des
séquences répétées et des
introns**. Ci-dessous l'exemple le virus
herpès humain 5 155573622.
Les gènes sont indiqués par des rectangles de
couleur verte ;
235.646
nucléotides, 79% d'ADN codant,
165 protéines et 2ARN non traduits, pas de
pseudogènes.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
*sens de transcription de l'ADN en ARN. En fait le complexe enzymatique (ARNpolymérase) qui polymérise les ribonucléotides triphosphate à partir d'un brin d'ADN qui lui sert de matrice, travaille toujours dans le même sens (3'->5') et donc (puisque l'ADN est constitué de deux brins antiparallèles, c'est-à-dire orientés dans deux sens opposés), lorsqu'il change de brin, il change automatiquement de sens. Lorsque deux gènes sont transcrits dans deux sens différents cela signifie qu'ils sont transcrits à partir de deux brins différents d'ADN.
**intron : séquence de l'ARN qui est excisée lors de la maturation (épissage) des ARN, principalement chez les eucaryotes. Par abus, on désigne par intron des séquences (de l'ADN) intragéniques non codantes (qui vont être transcrites en introns mais non traduites en protéines). Moyen mnémotechnique : intron = intercalé, exon = exprimé. |
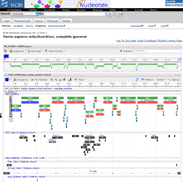
|
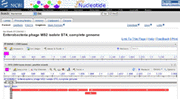
Voici le génome d'un petit
phage MS2 d'une entérobactérie: La fonction zoom maximal (+ à gauche) permet de visualiser les brins d'ADN, le triplet de bases de l'ADN correspondant à chaque codon de l'ARNm et les aa associés. Dans le fichier GenBank chaque portion d'ADN ou locus est décrite avec sa fonction et le produit qu'il code. Des hyperliens sont évidemment établis avec les séquences et les données existantes pour chaque produit. C'est un exemple de la qualité, mais aussi la complexité des résultats auxquels les biologistes moléculaires sont arrivés. |
|
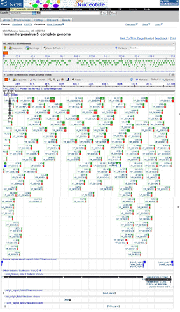
Pour comparer voici le génome d'un
phage lambda (10 fois plus grand)
cultivé sur une souche
d'entérobactérie |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
? De nombreux produits sont annotés, soit avec un point d'interrogation, soit comme "related" ou"hypothetical", soit enfin comme "putative" ce qui signifie, dans tous ces cas, que le produit est SUPPOSÉ, mais qu'il n'a pas été expérimentalement contrôlé (?) comme effectivement produit par le gène supposé. Ces suppositions se font à l'aide des programmes informatiques de reconnaissance automatique de séquence. Cela signifie donc seulement que ce gène présente une forte analogie de séquence avec un gène CONNU. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
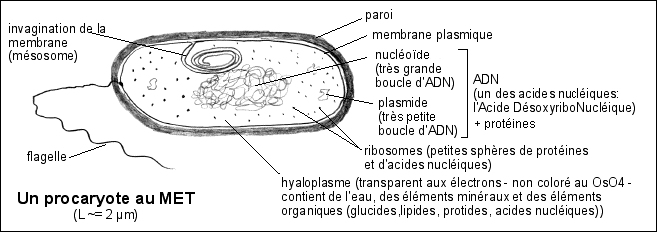
Du point de vue
structural : L'organisation de la cellule
procaryote a été vue en 2nde : Pour une image au MET voir par exemple le site de Markus Drechsler (E. coli au MET en coloration négative à l'uranylacétate). |
|
On considère ainsi que l'ADN est composé d'une grande boucle (appelée à tort parfois "chromosome bactérien") et de petits ADN circulaires: les plasmides. Mais chaque cellule procaryote contient plusieurs exemplaires de son ADN (environ 6 pour Escherichia coli). Quelques cas rares présentent un ADN double brin linéaire (Borrelia burgdorfei). Comme dans le cas des génomes linéaires viraux des protéines particulières sont observées au niveau de l'extrémité 5' de chaque brin.
|
|
Le nucléoïde est la structure qui contient l'ADN procaryote qui n'est ainsi pas réparti dans tout le cytoplasme. Pour une bactérie comme Escherichia coli, ce nucléoïde contient environ 40 % d'ADN (un peu d'ARN) et 60 % de protéines (non-histones: les protéines associées au nucléoïde sont d'une part les NAPs (Nucleoid Associated Proteins), ensuite les topoisomérases et enfin les protéines SMC (Structural Maintenance of Chromosomes, qui interviennent aussi chez les eucaryotes)). L'ADN procaryote n'est ni nu, ni libre, mais n'est pas non plus organisé en chromosome, pas plus qu'il ne possède de nucléosomes comme chez les eucaryotes. Cependant, on pense depuis quelques années que le nucléoïde bactérien est probablement aussi organisé en macrodomaines (à l'image de la chromatine chez la cellule eucaryote). La structure du nucléoïde bactérien n'est plus considérée comme unique et stable; et l'idée que sa dynamique puisse permettre le positionnement variable des différents domaines en fonction de l'âge et de l'état métabolique de la cellule est une hypothèse envisagée, même si l'on se refuse encore habituellement à considérer la position des domaines au sein du "chromosome" comme modifiable; il reste encore du chemin pour en venir à considérer que c'est la cellule qui, en fonction de son métabolisme, synthétise et positionne ses gènes. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
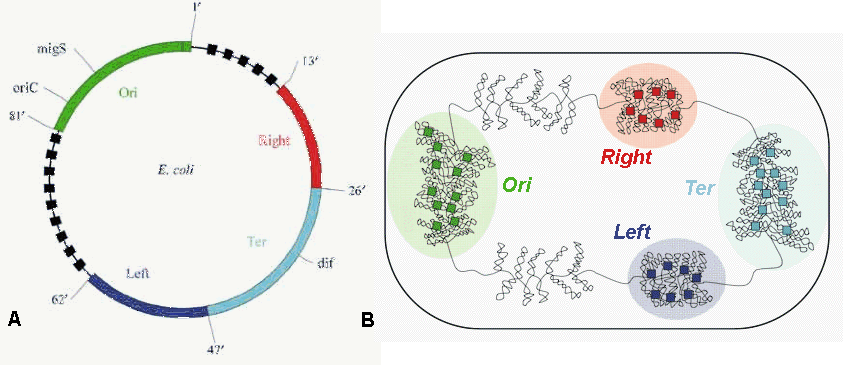
voir publications de l'équipe de Frédéric Boccard, CNRS
|
|
A. Le cercle représente la carte génétique du "chromosome". Les barres colorées représentent les différents macrodomaines (Ori, Right, Ter et Left) et les barres noires interrompues schématisent 2 régions moins structurées. L'origine de réplication oriC, les sites migS et dif sont indiqués. B. Un modèle pour l'organisation
du "chromosome" chez E. coli où des supertours de
l'hélice d'ADN sont figurés. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
L'ADN procaryote est attaché, au moins pendant la phase de division cellulaire, à la membrane cellulaire et/ou à la paroi. La théorie de la duplication de
l'ADN de la cellule procaryote au cours d'un cycle
cellulaire a été élaborée
à partir des expériences
historiques de Meselsohn et Stahl en
1958. Cependant, il reste encore
bien des points mystérieux, notamment du fait de la
taille des bactéries et donc des difficultés
d'observation (voir page
présentant cette
publication).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
La base de données des
génomes procaryotes du NCBI contient un très
grand nombre de génomes cartographiés : |
|
Une cyanobactérie
(Acaryochloris marina MBIC11017) avec un génome
de 6.503.724
nucléotides dans
GenBank
158303474. |
|
Escherichia coli souche
BL21(DE3 ) |
|
Lactobacillus casei
(ATCC334) : Remarque: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Du point de vue fonctionnel Expression |
|
Stabilité et transmission |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
L'ADN procaryote contient des gènes organisés linéairement SANS INTRONS sauf très rares exceptions (intron est employé ici abusivement pour désigner de l'ADN non codant intragénique transcrit en intron*). Mais il existe de l'ADN intergénique (typiquement 10%). La quasi totalité de l'ADN génique du procaryote est codant ou -autrement dit - est transcrit en ARN, puis éventuellement traduit en protéines. Mais qu'en est-il de l'ADN intergénique ? Il semble qu'il ne soit pas transcrit (il est donc a fortiori non codant). Mais cela est difficile à démontrer et l'on est pas sûr de ne pas se tromper. Il pourrait être transcrit sans donner de produit fonctionnel, comme cela est le cas pour les "introns" des eucaryotes par exemple. Certains gènes sont chevauchants.
Chaque brin peut être transcrit
séparément; la transcription du même
locus dans des sens différents (et donc sur des brins
différents) donnant des produits différents
... Bref, l'expression de l'information
génétique procaryote est complexe. |
|
Il est important de comprendre que notre
vision du fonctionnement cellulaire comme résultat de
l'expression d'une information génétique (le
fameux programme génétique) vient pour
une grande part du modèle bactérien. Toute
nouveauté dans le modèle devrait nous inciter
à remettre en question sa pertinence.
|
|
Jusqu'à peu on considérait que l'ADN était stable et transmis à deux bactéries-filles, de façon idéalement identique, après une reproduction conforme, aux erreurs près, en phase S de croissance du cycle cellulaire (voir cours). Le point central était donc qu'un génome (par analogie à la vie !!!) était une unité qui pouvait être dupliquée et transmise mais non pas fabriquée de novo. Le dogme était « la cellule ne fabrique pas ses gènes mais en hérite, les duplique et les mute ». Ainsi seuls trois types de changements
étaient considérés comme possibles
: Depuis que l'on a découvert chez
un grand nombre d'organismes des systèmes
enzymatiques de transcription inverse, il n'est plus
aussi certain que ce soit l'ADN qui représente la
forme la plus stable d'une information
génétique. |
|
Depuis le début de la mise en
place du paradigme de l'information génétique
déterministe et stable, il n'a pas manqué de
voix pour présenter des travaux qui allaient dans un
sens différent (voir la
discussion
dans la page sur les
mutations). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Un fragment de génome mais sans redondance dans le génome nucléaire ...
... des compartiments cellulaires pour un métabolisme spécifique et pourvu d'un matériel génétique spécifique en rien autonome. |
|
L'appellation "génome" pour les organites est tout aussi usurpée que pour les virus et peut mener à un préjugé erroné d'une composition du "génome" suffisante pour expliquer les éléments d'un organite autonome (selon la théorie de "l'information génétique"). Or un organite n'est pas un organisme et n'est donc pas autonome (même si une théorie dite endosymbiotique propose de les faire dériver de procaryotes symbiotes de la cellule eucaryote - voir remarque ci-dessous). Son "génome" code pour des produits utilisés dans l'organite lui-même mais aussi pour des produits utilisés autre-part dans la cellule. Enfin la plupart des protéines nécessaires à la croissance (et à la division) de ces organites sont synthétisées à partir de gènes nucléaires. Le "génome" de l'organite n'est donc qu'un petite partie du génome de l'organisme. Le "génome" des organites est toujours composé d'ADN double brin (généralement circulaire mais des formes linéarisées sont connues). Chaque organite contient de multiples copies de son ADN. Certains gènes contiennent des introns. Aucun des gènes du "génome" de l'organite ne se trouve au sein du génome nucléaire (il n'y a pas de redondance). Il est à noter que, si chaque organite possède des ribosomes, l'activité de transcription-traduction au sein de leur matrice reste très réduite. Les complexes enzymatiques de type ARNpolymérase sont d'origine strictement nucléaires, mais peuvent bien sûr être hérités. |
|
Donc la question du sens du "génome" de l'organite reste entière. Peut-être est-ce justement l'occasion de proposer d'autres rôles au "génome", puisque qu'il n'est pas utilisé pour accéder à une information de structure ni même de fonction. Pourquoi une information pour quelques protéines et ARN utilisés dans l'organite (ou pas pour certains, voir ci-dessous) et pourquoi ces molécules ? Si la composition de ce "génome" ne peut pas être comprise à partir de son rôle informatif dans les synthèses (ARN et protéines), on ne peut qu'imaginer des mécanismes de structuration en rapport avec la dynamique des organites eux-mêmes. Après tout, le chondriome, par exemple, n'est qu'un compartiment dynamique supplémentaire qui peut très bien être mis en balance avec le compartiment du reticulum qu'est le noyau. La ségrégation d'un "génome" en leur sein pourrait résulter du même type de dynamique.
|
|
La dynamique de ces organites est une donnée récente et reste à explorer. Cela est particulièrement vrai pour les mitochondries, qui sont plutôt organisées en chondriome (tubules mitochondriaux anastomosés) dans de nombreuses cellules. Certains films montrent bien ce passionnant phénomène (voir ci-contre et cours de seconde). Les chloroplastes ne sont pas à l'abri de ces remaniements ; on sait depuis fort longtemps qu'ils sont susceptibles de se diviser plus ou moins activement, de se transformer en diminuant ou en augmentant le nombre de leurs thylakoïdes et grana, et enfin de se spécialiser (amyloplastes) au cours de la vie de la cellule qui les renferme. La plupart des cellules meurent lorsqu'elles perdent leur mitochondries et/ou leur chloroplastes mais le nombre de ces organites par cellule varie grandement en fonction de l'âge ou du métabolisme cellulaire. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
On peut comparer le "génome" mitochondrial d'Homo sapiens et d'H. sapiens neanderthalensis |
|
Homo sapiens - "génome"
mitochondrial complet (251831106) : ADN circulaire double
brin, 16.569
nucléotides, 37 gènes
(13 de protéines et 24 d'ARN (2ARNr, 22ARNt)), 68%
d'ADN codant
|
|
Le protéome de la mitochondrie
humaine (13 gènes): |
|
L'intérêt des études sur le "génome mitochondrial" repose pour une grande part sur la recherche médicale concernant la transmission strictement maternelle des gènes (moléculaires) mitochondriaux (et donc de gènes héréditaires qui seraient associés - voir chapeaux encadrés du cours pour cette distinction). En effet, on pense que, lors de la fécondation, aucune mitochondrie du spermatozoïde de l'homme ne pénètre dans le cytoplasme de l'ovocyte féminin. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Les chiffres sont ceux des bases de données mais correspondent, la plupart du temps, à une synthèse de nombreux résultats, plus ou moins cohérents; l'ADN mitochondrial n'est pas toujours double brin, ni circulaire, et parfois très fragmentaire ou répétitif dans un organite donné... Il va sans dire que le "génome" mitochondrial peut être fort variable entre cellules et même entre mitochondries d'une même cellule chez un organisme. |
|
Diversité des "génomes"
mitochondriaux
(j'ai
volontairement choisi quelques-uns qui
s'écartaient du modèle humain pour
donner un panorama de la
diversité)
protistes
(30 taxons connus
environ)
Paramecium aurelia
(une paramécie
autogame)
Plasmodium falciparum
(parasite, agent du
paludisme)
Plasmodium
juxtanucleare
Dictyostelium discoideum
(amibe acrasiale)
champignons
(près de 1900
taxons connus en comptant les animaux)
Aspergillus niger
(moisissure)
Candida orthopsilosis
(rare, "génome"
mitochondrial de Candida albicans non
connu)
Rhizopus
oryzae
(moisissure)
Saccharomyces
cerevisiae
Ustilago maydis
(charbon du
maïs)
plantes
(50 taxons connus
environ)
Chondrus crispus
(algue rouge)
Fucus vesiculosus
(algue brune)
Marchantia polymorpha
(hépatique
à thalle)
Nicotiana tabacum
(le tabac)
Arabidopsis
thaliana
animaux
(près de 1900
taxons connus en comptant les champignons)
Anacropora matthai
(corail)
Heterorhabditis
bacteriophora (ver
nematode)
Taenia solium
(ver solitaire du
porc)
Architeuthis dux
(céphalopode
géant)
Anopheles funestus
(moustique)
Anopheles gambiae
(moustique transmettenat
le paludisme)
Alligator
mississippiensis
Apus apus
(hirondelle)
Canis lupus
(loup gris)
Homo
sapiens
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
Un très grand nombre d'animaux
étudiés possède un "génome"
mitochondrial très voisin de celui de l'homme. Mais
il y a des exceptions. Les plantes étudiées (très peu nombreuses) ont clairement un "génome" mitochondrial plus grand avec parfois un assez grand nombre de protéines codées (dont la plupart restent hypothétiques). Les protistes présentent la plus forte diversité des "génomes" mitochondriaux. |
|
On a donc l'impression très nette que l'interprétation de la variabilité repose directement sur l'amplitude de la connaissance. Cette dernière est-elle suffisante pour avoir une vue d'ensemble ? J'en doute. De plus, il existe un raisonnement trompeur. Rien n'est plus simple comme idée que de proposer la transmission d'un "génome" ancestral puis la perte (ou le gain) de certains gènes dans différents phylums. Cette vision simpliste, que l'on pourrait qualifier de moléculariste tout autant qu'évolutionniste synthétique (en référence à la théorie synthétique de l'évolution) , n'est pas étayée par l'observation, mais reste une grille de lecture. On peut douter de sa pertinence tant que l'on n'a pas progressé sur la signification métabolique du "génome" mitochondrial. |
|
Les gènes d'ARN sont évidemment limités en type et il n'est pas surprenant qu'ils soient absents (0) ou présentant la panoplie assez complète d'ARNt (22), à quelques unités près. Plus surprenant, le fait qu'ils soient réduits à 4 ARNt (chez Anacropora matthai) et avec 4 ARNr (chez Paramecium aurelia). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Les mutants "petite" de la
levure de bière sont des souches chez qui on observe
des lésions plus ou moins importantes dans l'ADN
mitochondrial (voire même l'absence d'ADN). Leur nom
provient du fait que les cellules restent toujours de petite
taille en culture. Ces mutants apparaissent
spontanément avec une fréquence de l'ordre de
1% des colonies. Pour sélectionner les souches que
l'on considère comme de phénotype "petite" on
utilise un milieu sélectif où les cellules ne
peuvent se développer sans respiration
(10g d'extrait de levure, 0,25g de
glucose, 25 mL de glycérol, 20g d'agar, 980mL
d'eau / à comparer avec un milieu complet contenant,
en moins le glycérol, et en plus 2-30g d'extrait de
levures, ± bactopeptone ± extrait de
malt). On considère que le
glycérol ne pouvant être fermenté les
cellules "petite" ne se développent pas
(TP de biologie des levures, Didier
Pol, Ellipses).
|
|
Une fois encore la méthode sélective est bien incapable de prouvez quoi que ce soit au sujet d'une relation entre le génotype et le phénotype (voir page sur les mutations pour un approfondissement). Que les souches "petite" sélectionnées présentent des altérations de leur ADN mitochondrial ne prouve pas que ce sont ces altérations qui sont la cause du phénotype. On est même en droit de penser qu'au contraire leur modification métabolique est la cause de leur déficit en ADN. Mais on observe un autre phénomène qui ne va pas non plus dans le sens d'une causalité génotype mitochondrial -> phénotype. Les études fonctionnelles réalisées sur les mitochondries de souche "petite" , même celles présentant une régression de leurs crêtes, ont montré que les éléments de la chaîne respiratoire sont presque au complet et que les mitochondries semblent fonctionnelles. On a souvent interprété ce fait par l'écrasante domination du génome nucléaire pouvant pallier une déficience mitochondriale (Biologie moléculaire de la cellule, Alberts et al. Flammarion-Médecine-Sciences, p 713). Mais on s'est bien gardé de remettre en question le modèle déterministe génotype -> phénotype indûment appliqué ici à la mitochondrie. |
|
Il est évident que cette
interprétation est loin d'être la seule
possible. Il ne faut pas oublier que l'on ne sait pas
comment la cellule "fabrique" ses mitochondries: toute
mitochondrie étant héritée. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Les chiffres sont ceux des bases de données mais correspondent, la plupart du temps, à une synthèse de nombreux résultats, plus ou moins cohérents; l'ADN plastidial n'est pas toujours double brin, ni circulaire, et parfois très fragmentaire ou répétitif dans un organite donné... Il va sans dire que le "génome" plastidial est fort variable entre cellules et même entre chloroplastes d'une même cellule chez un organisme. |
|
Diversité des "génomes"
chloroplastiques
Euglena gracilis
Chlorella vulgaris
Porphyra purpurea
(algue rouge)
Chara vulgaris
("algue"
characée)
Marchantia polymorpha
(hépatique
à thalle)
Cycas taitungensis
Brachypodium distachyon
(Graminée)
Hordeum vulgare
(orge commun,
Graminée)
Saccharum officinarum
(canne à
sucre)
Zea mays
(le maïs)
Epifagus virginiana
(parasite Orobranche)
Pinus koraiensis
(un pin rare
coréen)
Pinus contorta
Daucus
carota (la
carotte)
Nicotiana tabacum
(le tabac)
Platanus
occidentalis
Arabidopsis
thaliana
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
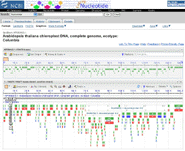
|
Le "génome" chloroplastique de la plante modèle Arabidospsis thaliana n'est pas particulièrement représentatif mais ne constitue pas non plus une exception; il est détaillé ci-dessous. C'est l'Orobranche parasite Epifagus qui présente le "génome" le plus réduit. Là encore, trop peu de taxons sont connus (170 dans la base NCBI) pour que l'on puisse dégager des traits généraux. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Une diversité importante mais des synthèses très limitées |
|
Arabidopsis
thaliana a un "génome"
chloroplastique de
154.478
nucléotides sous la forme d'un
ADN circulaire double brin (mais, là encore, des
formes linéaires sont connues, et il y a une grande
variabilité de matériel entre les organites:
le "génome" circulaire présenté est en
quelque sorte une représentation moyenne
théorique qui ne tient notamment pas compte des
nombreux exemplaires de chaque gène...). |
|
ADN d'un chloroplaste d'A. thaliana ;
représentation graphique dans la base GenBank
: |
|
En comparaison du "génome" mitochondrial le "génome" chloroplastique est incontestablement plus riche mais sans que l'on aie une structure différente : 7 ARNr (2x16S, 2x23S, 2x4,5S, 5S) et 37 ARNt (dont certains en double). Les protéines codées par les gènes chloroplastiques appartiennent à la chaîne photosynthétique, on retrouve aussi des sous-unités de l'ATPsynthase, mais aussi des protéines ribosomales, ou encore des sous-unités de l'ARNpolymérase, absentes dans la mitochondrie... |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Chloroplastes sans ADN... |
|
Les lignées d'Euglènes présentant une régression chloroplastique, une fois cultivées à l'obscurité, sont parfois interprétées avec le même type de raisonnement simpliste que celui réalisé pour les colonies "petite" de levure de bière (Biologie moléculaire de la cellule, Alberts et al., Flammarion-Médecine science, 713). |
|
On notera que chez Acetabularia près de 50% des plastes ne contiennent pas d'ADN. L'interprétation actuelle, bancale, est qu'il existerait une incompatibilité génomique entre les "génomes" nucléaire et plastidial (voir page spéciale). Plutôt que de remettre en cause le modèle on fait intervenir un génome nucléaire à la rescousse pour expliquer les déficiences du "génome" plastidial. C'est encore une fois abuser de la comparaison entre organite et organisme. |
|
L'organite ne représente qu'une toute petite part du métabolisme (une région très limitée de l'espace des régulations métaboliques de la cellule) et rien n'empêche que l'on y observe des zones de discontinuité métabolique conduisant à une absence de synthèse d'ADN. Ce qui tend d'ailleurs à proposer de séparer la genèse de la forme de l'organite avec la synthèse d'ADN qui est associée à son fonctionnement. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
Remarque: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
Générer des
structures (boules, bâtonnets, tubes
anastomosés...) par des plissements de l'espace
métabolique des régulations. On notera que la
genèse de ces formes pourrait être identique
entre des unicellulaires procaryotes et des organites
eucaryotes (ce sont en effet les mêmes formes). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
1.4.1 - L'ordre à l'échelle
de la chromatine et du chromosome
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
1.4.1.1 - l'ADN
nucléaire n'est pas nu et parfois très
condensé grâce à des protéines
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
À l'exception de l'ADN mitochondrial et plastidial (voir ci-dessus), l'ADN des cellules eucaryotes est compacté (séquestré) , en dehors des phases de division, au sein d'un organite : le noyau, que l'on peut considérer comme une citerne délimitée par la double membrane du réticulum endoplasmique (percée de pores). Il y a donc un lien fort entre synthèse d'ADN et formation et maintien de la forme du réseau endoplasmique (dynamique). |
|
Lorsque la cellule est au repos (et non en phase M de son cycle cellulaire) le contenu nucléaire, nommé chromatine, se présente comme une masse plus ou moins homogène, basophile (qui prend facilement les colorants basiques), où l'on peut distinguer un certains nombre de nucléoles (zones ayant une affinité encore plus marquée pour les colorants basiques).
|
|
La chromatine contient environ 50% d'ADN et 50% de protéines. Au microscope électronique
à transmission on peut distinguer deux types de
chromatine : Le modèle habituellement retenu
est celui d'une fibre chromatinienne unique, plus ou moins
condensée. |
|
La structure des
nucléosomes est connue. Ils sont
composés de sous-unités de protéines du
groupe de histones, qui sont les protéines basiques
responsables de colorations de la chromatine sur les
préparations observées au MO. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
En travaux |
|
Nouvelles données sur les nucléosomes BMG |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
En phase de division (M) l'enveloppe nucléaire se fragmente (chez la plupart des organismes, mais pas chez de nombreux unicellulaires) et le contenu nucléaire apparaît sous forme de chromosomes (masses allongées colorables au MO et au ME) qui se déplacent dans la cellule, se fragmentent, puis sont repartis dans les cellules filles issues de la division cellulaire (voir mitose). |
|
Le chromosome contient davantage de protéines que la chromatine : environ 1/3 d'ADN pour 2/3 de protéines (pour moitié basiques, de type histones, pour une autre moitié, acides, non histones). On considère habituellement que le chromosome est issu de la condensation de la chromatine, mais la structure du chromosome reste très hypothétique. Il reste fondamentalement à expliquer l'origine du doublement de la quantité de protéines entre la chromatine et le chromosome. Le problème de l'accessibilité de l'ADN par la machinerie métabolique qui assure son expression (sous forme d'ARN par transcription) n'est pas un problème résolu. Certains gènes sont clairement transcrits lors de la phase de division. Des hypothèses de décondensation partielle ont été émises, à l'instar des chromosomes "en écouvillon" que l'on observe dans des cellules qui présentent une forte activité de synthèse protéique (ovocyte II des amphibiens par exemple). |
|
Cependant, tout l'ADN ne se condense pas en chromosomes. En effet, le ou les nucléoles restent souvent séparés et sont plus ou moins répartis entre les cellules filles. Il existe aussi une certaine quantité d'ADN extrachromosomique chez certaines espèces. Il semble que cet ADN contienne surtout des gènes codant pour des ARN ribosomiaux. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
L'ADN nucléaire n'est pas
composé d'une seule molécule
(3.000.000.000 de nucléotides
chez l'homme ce qui correspond à environ 1 m d'ADN on
comptant 10,5 pb par tour et 3,4 nm par tour
d'hélice) mais
fragmenté. Lors de la division (mitose) l'ADN est
principalement présent dans les chromosomes, ce qui
rend évident sa fragmentation. Lors de la phase de
repos (interphase) il existe aussi une territorialisation
qui peut faire penser que cette fragmentation persiste.
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
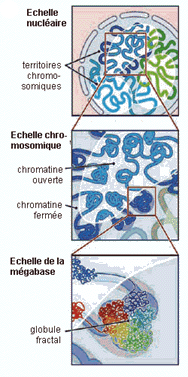
Des territoires nucléaires chromosomiques... |
|
Selon l'interprétation la plus habituelle chaque chromosome apparaissant lors de la mitose correspond à une unique et gigantesque molécule d'ADN. Chaque cellule aurait ainsi le même nombre de molécules d'ADN que de chromosomes (à la différence près, bien sûr, qu'un chromosome prophasique à 2 chromatides contiendrait 2 molécules d'ADN identiques répliquées lors de la phase S du cycle cellulaire). Voilà pour le "dogme". Mais les choses sont sans aucun doute plus complexes. |
|
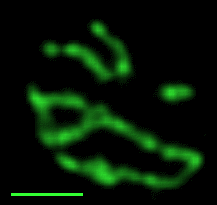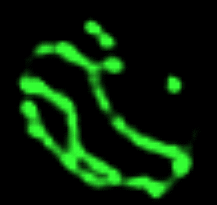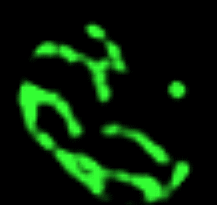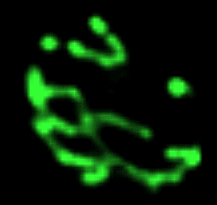
On a effectivement reconnu, notamment à l'aide des systèmes immunofluorescents de marquage chromosomique, que chaque chromosome condensé correspondait bien a un territoire nucléaire. simulation |
|
Mais ce n'est pas pour autant que l'on sait comment on passe d'un état à l'autre et surtout comment se présente et s'organise l'ADN à chaque niveau. Les chromosomes correspondent sans aucun doute à des territoires nucléaires associés à un paysage métabolique qui pourrait donc être mis en relation avec une région de l'espace des régulations. Chaque chromosome serait la trace d'une région de l'espace métabolique. La structuration de l'ADN serait alors la signature du type de métabolisme associé à chaque paysage. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Repliement de la fibre chromatinienne qui pourrait se condenser en chromosome lors de la division |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Un exemple de mélange des techniques.... un travail colossal d'une nombreuse équipe alliant biochimie, technique de séquençage massivement parallèle et outils statistiques... pour un résultat pas si convaincant
N.B. le terme "chromosome" est souvent employé de façon abusive pour désigner la chromatine ou fibre chromatinienne... |
|
Les études les plus
récentes concernant l'organisation nucléaire
du génome tentent de concilier les méthodes de
la métagénomique avec la morphologie. Voici
quelques éléments d'un article récent
qui me paraît représentatif: |
|
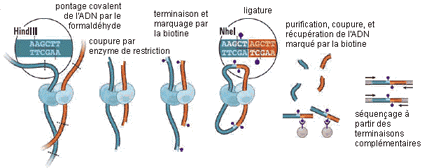
L'objectif : La méthode (Hi-C) : « Here, we
report a method called Hi-C that adapts the above
approach to enable purification of ligation
products followed by massively parallel sequencing.
Hi-C allows unbiased identification of chromatin
interactions across an entire genome.We briefly
summarize the process: cells are crosslinked with
formaldehyde; DNA is digested with a restriction
enzyme that leaves a 5! overhang; the 5! overhang
is filled, including a biotinylated residue; and
the resulting blunt-end fragments are ligated under
dilute conditions that favor ligation events
between the cross-linked DNA fragments. The
resulting DNA sample contains ligation products
consisting of fragments that were originally in
close spatial proximity in the nucleus, marked with
biotin at the junction. *
méthode avec dépôt de brevet en
cours.....
Nous
présentons ici une méthode*
nommée Hi-C qui modifie l'approche
précédente afin de permettre la
purification de produits liés ainsi qu'un
séquençage massivement
parrallèle. Hi-C permet d'identifier de
façon impartiale les interactions de la
chromatine au niveau du génome entier.
Résumons brièvement le
procédé: les cellules sont
fixées avec le formaldéhyde ; l'ADN
est digéré par une enzyme de
restriction qui laisse un bout;collant 5'; le
bout-collant 5' est complété et un
résidu biotinylé est ajouté ;
et les fragments à bouts francs sont
ligaturés dans des conditions de dilution
qui favorisent la liaison entre les fragments d'ADN
pontés. L'échantillon d'ADN qui en
résulte contient donc des
éléments ligaturés qui
étaient situés auparavant dans le
même voisinage au sein du noyau, et qui sont
maintenant marqués par de la biotine au
niveau de leur jonction. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
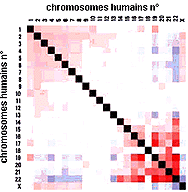
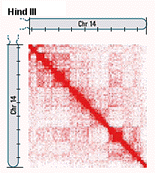
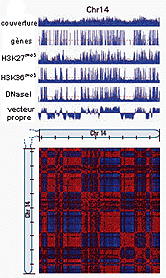
Résultats : Ce résultat n'est pas nouveau mais sa cohérence avec les modèles actuels fournit un argument en faveur de la validité de la méthode Hi-C. On notera ainsi que les petits chromosomes riches en gènes (n° 16, 17, 19, 20, 21 et 22) interagissent préférentiellement entre eux selon le modèle obtenu à partir de la FISH, ce qui est cohérent avec l'Hi-C. On notera aussi que le chromosome 18, petit mais pauvre en gènes n'interagit pas fréquemment avec les autres chromosomes, ce qui est aussi concordant avec le modèle obtenu par FISH qui le place à la périphérie du noyau. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
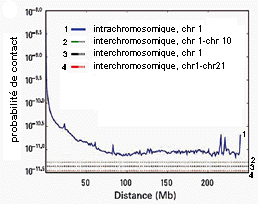
Au sein d'un même chromosome les
interactions sont mesurées en fonction de la taille
des fragments considérés. Une fois encore les
résultats confortent les données obtenues par
FISH. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
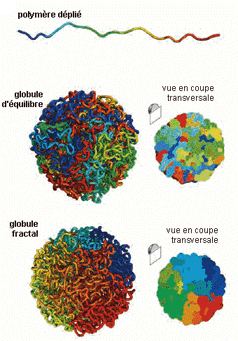
Un traitement mathématique relativement complexe est ensuite utilisé pour interpréter les résultats afin de supprimer l'effet de proximité entre deux loci (les résultats sont normalisés par rapport à une matrice qui ne tient compte que de la distance); puis une analyse en composantes principales permet de proposer une partition de chaque chromosome en domaines de deux types : chromatine ouverte (open) et chromatine fermée (closed).
|
|
Modèle : |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
« Of
course we cannot rule out the possibility that
other forms of regular organization might lead to
similar findings.»
« Bien
sûr nous ne pouvons exclure la
possibilité que d'autres formes
d'organisation régulière puisse mener
à des résultats
identiques».
C'est tout le problème
de ces modèles, certes cohérents avec
les résultats, mais il existe un grand
nombre de formes qui pourraient donner des
résultats identiques, à commencer par
des boucles d'ADN courtes et non pas une unique
molécule de dimension faramineuse. Les
techniques mises en jeu ici exigent une confiance
très forte dans le modèle initial
pour que les résultats puissent être
interprétés dans un sens
cohérent. La question de savoir si l'on a
pas construit un château de cartes n'est pas
une question oiseuse mais un véritable
problème de signification en biologie
moléculaire : voir page suivante pour un
approche des techniques. Les points les plus
délicats restent le rôle réel
du formol (technique d'immunoprécipitation
de la chromatine ou ChIP) et toutes les
étapes de coupure-ligation-extraction
jusqu'au séquençage au hasard
(shotgun sequencing).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
1.4.2 - Le désordre à l'échelle des fonctions : gènes et autres séquences On ne répétera jamais assez que le gène n'est pas défini par sa structure mais par sa fonction. Trop souvent on parle de gène de façon hypothétique sans avoir vérifié in vivo qu'une séquence correspondait effectivement a un produit. Sans produit (ARN et/ou peptide) il n'y a pas de gène. Les études in silico ont multiplié les gènes hypothétiques et ont conduit à brouiller notre compréhension de la structure et surtout de la dynamique du génome. Ceux qui travaillent sur des réseaux effectifs de gènes savent combien est complexe la dynamique des fonctions faisant intervenir les gènes et leurs produits. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
1.4.2.1 - L'ADN nucléaire est plus ou moins
pauvre en gènes
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Des chiffres qui reposent sur des estimations |
|
Génomes
nucléaires de quelques espèces-modèle
eucaryotes
|
|
* les estimations du nombre de gènes varient fortement d'une source à l'autre, principalement parce que la plupart des gènes estimés sont uniquement supposés à partir de la reconnaissance automatique de similitudes de séquence avec des gènes connus, et ensuite parce que un certain nombre de gènes sont dupliqués, parfois un grand nombre de fois. Séquençage de
chromosomes
eucaryotes complets |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
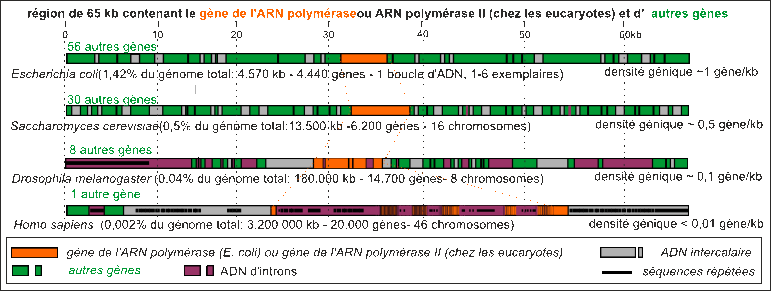
UNE
BELLE PAGAILLE !
Comparaison de la
densité
génique de
l'ADN de 4 organismes au voisinage du gène
de l'ARNpolymérase (une région de
65kb est
représentée). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Le nombre de gènes nucléaires varie entre 5.000 et 50.000 sans rapport avec la complexité des organismes |
|
Si l'on utilise le même paramètre (nombre de gènes/taille du génome; ce qui ne tient pas compte bien sûr des gènes chevauchants...) la densité génique des virus est de l'ordre de 1 à 1,5 gène/kb et celle d'une mitochondrie humaine 2 gènes/kb... De telles valeurs indiquent peut-être un changement de signification de la répartition des gènes au sein de l'ADN pour ces "génomes". Cela conforte l'idée que ces fragments d'ADN NE SONT PAS DES GÉNOMES. La fabrication de ces acides nucléiques n'est peut-être pas réalisée par le même type de mécanisme que celui des cellules vivantes. |
|
Dans le cas d'organismes vivants - et les procaryotes sont probablement à classer à part -, la complexité de la structure du génome reflète la complexité de l'organisme et des dynamiques GLOBALES, et non pas seulement cellulaires (voir aussi tableau sur fond jaune ci-dessus). On pourrait penser que plus un
organisme est complexe plus il devrait posséder de
gènes, mais ce n'est pas ce que l'on observe au
niveau cellulaire : le nombre de
gènes par cellule n'est pas si différent
entre organismes (les procaryotes mis à part avec
de l'ordre de 500 à 6.000 gènes) avec des
chiffres estimés entre 5.000 et 50.000
gènes. Leur nombre oscille entre un facteur 1
à 10 selon les organismes, mais indépendamment
de la taille ou de la complexité des organismes.
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
La première hypothèse, la plus évidente, serait alors que le morcellement augmente lors de chaque division. Plus un organisme possède de cellules plus son ADN serait fragmenté. Cela reste à démontrer. Corrélativement il ne semble pas (même si l'exploration précise n'a sans doute jamais été faite) qu'entre une cellule embryonnaire et une cellule différenciée la masse d'ADN soit si différente, du moins de façon constante et avérée (on rapporte de nombreux cas de polyploïdies ou au contraire d'ADN manquant, dans les cellules différenciées, mais aucune généralisation ne peut être faite actuellement). |
|
Une autre idée séduisante reste celle de l'évolution. les organismes les plus simples étant supposés être apparus d'abord, la complexité des génomes reflétant alors l'évolution des organismes. Mais, attention, il ne s'agit pas ici d'évolution par ajout ou modification de gènes, mais bien par ajout des séquences intercalaires et/ou répétées... Il est clair que ce ne sont pas les organismes les plus évolués (au sens de distance phylogénétique maximale et non d'éloignement dans le temps) qui ont le plus de gènes. |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
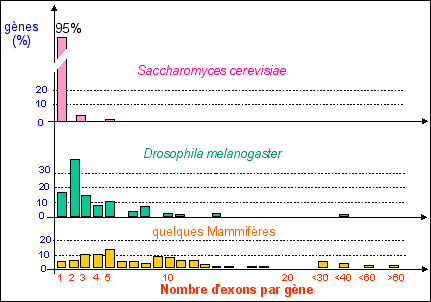
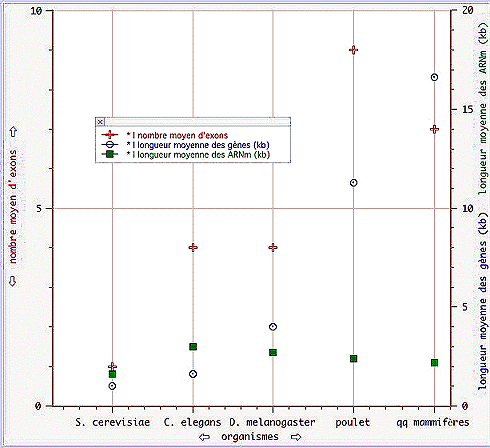
|
le morcellement des gènes en exons-introns |
|
La découverte des introns désormais classique repose sur la comparaison (par réalisation d'un hybride ADN-ARN) entre la longueur du gène transcrit et la longueur de l'ARNm traduit. Ce dernier étant sensiblement plus court dans de très nombreux cas. On nomme introns les séquences de l'ARN (issues de la transcription d'un gène) non codantes qui sont, la plupart du temps excisés, c'est-à-dire coupées, lors de la maturation de l'ARNmet avant sa traduction au sein des ribosomes. Les exons sont alors les séquences codantes du transcrit primaire qui, bout-à-bout, composent l'ARNm. La répartition et le rôle des introns reste un mystère. |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
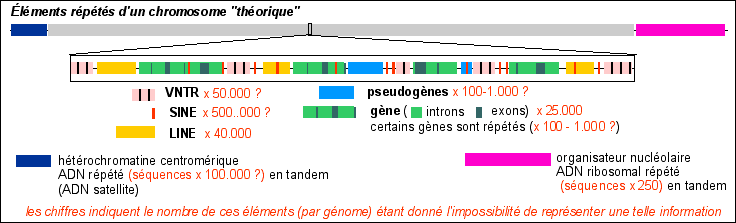
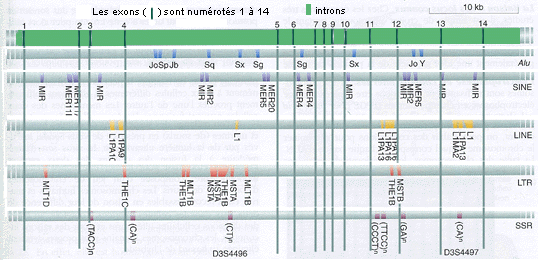
Éléments répétés à deux échelles Pour se rendre compte de la signification des éléments répétés (voir plus bas pour les sigles) à deux échelles différentes voici deux représentations fort éloignées: |
|
À l'échelle
d'un chromosome : À l'échelle
d'un gène
: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
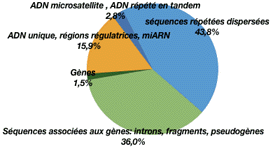
Les éléments répétés correspondent à près de 50% du génome mais l'on ne tient pas compte des gènes répétés |
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Un ensemble très hétérogène
|
|
séquences répétées des
télomères |
|
séquences répétées des
centromères |
|
Taille et composition de quelques centromères (in BMG fig 7.8) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
O-O-O-O-O-O-O O.....O-O........O...O............O |
|
origines de réplication
répétées séquences répétées
péricentromériques = ADN satellite |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
* les répétitions en tandem en nombre variable (variable number tandem repeats = VNTR) ou ADN minisatellite. Leur fonction est inconnue. |
|
* ADN microsatellite = régions dispersées composées d'un nombre variable de dinucléotides répétées en tandem. Cet ADN a fourni de nombreux marqueurs moléculaires pour la cartographie de génomes de grande taille. Leur fonction est inconnue. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
gènes répétés |
|
organisateur nucléolaire |
|
ARNt histones |
|
pseudogènes |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
séquences mobiles (répétées) elles correspondent à des séquences d'ADN qui sont fortement apparentées à de nombreux provirus (ch 13 AGM); ces séquences peuvent représenter la majorité de la masse d'ADN répété d'une cellule. |
|
transposons rétrotransposons |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ADN intercalaire s.s. |
|
L'ADN intercalaire (spacer en anglais) correspond en gros à l'ADN dans lequel on a pas pu identifier ni gène, ni éléments répété, ni élément mobile.... C'est de l'ADN en attente de compréhension... |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
génome de Cænorhabditis elegans
Source: |
|
Premier génome métazoaire séquencé en 1998. Chromosomes sans centromères (fait rare mais pas unique) mais avec des télomères classiques (de séquence TTAGGC). Nombreuses séquences répétées dispersées entre les gènes et dont la fréquence augmente le long des bras des autosomes. 2,7% de régions répétées en tandem, 3,6% de régions répétées et inversées. Il existe aussi des régions répétées dispersées correspondant en majorité à des éléments transposables défectifs. Chaque chromosome (sauf le sexuel X) comporte une polarité qui apparaît lorsque l'on analyse la distribution de petites séquences répétées (MITE: miniature inverted-repeat transposable element); ces courtes séquences présentant une distribution asymétrique le long des chromosomes. Grande variété de transposons de type bactérien ainsi que plusieurs sortes de rétrotransposons. |
|
L'analyse automatique par reconnaissance de séquence donne environ 20.000 gènes codants pour des protéines; les exons représentant 27% du génome et les introns 14%. Plusieurs centaines d'autres gènes: 659 ARNt, ARNs(U1 à U6 formant le spliceosome ), ARNs leader (SL1 et SL2), plus des ARNr, ARNtélomériques et des snARNs. Plusieurs dizaines de gènes forment de petits ARNs non traduits.
|
|
Les analyses semi-automatiques des
protéines ont permis de reconnaître 20 familles
qui représentent 3.000 gènes (650 reconnus par
exemple pour les chimiorécepteurs, 410 domaines de
type protéine kinase, 240 domaines en doigt de zinc
de type C2-C2, 170 domaines de type
collagène...). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
en travaux |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
génome de l'homme |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ADN-ARN en interaction |
|
ADN et ARN sont hérités
avec : Il est loin le temps où l'on pensait que le seul matériel génétique à être transmis était l'ADN. |
|
Si on continue à n'enseigner le plus souvent que les seules hérédités chromosomique et mitochondriale (et chloroplastique), les travaux qui rétablissent l'importance de l'hérédité cytoplasmique sont nombreux. |
|
Mais étant donné la difficulté de prouver expérimentalement ce type d'hérédité, tout comme l'extrême difficulté à établir des modifications génomiques qui auraient lieu lors du développement ou lors du vieillissement, on se contente souvent du modèle ancien alors que tout chercheur sait pertinemment que l'ADN (eucaryote) provient (probablement) au moins autant de l'ARN que de l'ADN par héritage entre cellules. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
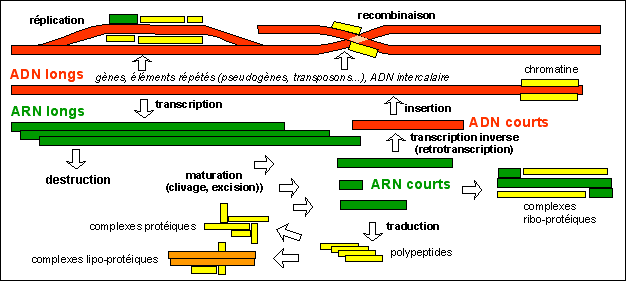
|
une molécule unique ou un amas de molécules d'origine variée ?
ADN
->
ARN
(longs et
courts)
|
|
Comment concilier les résultats de la génétique moléculaire et un modèle qui ne considère pas que chaque chromosome possède une unique molécule d'ADN stable et transmise par hérédité ? Tant que l'on aura pas une idée précise du métabolisme de l'ADN, on ne pourra pas comprendre l'organisation nucléaire. Que l'on ait de petits ADN circulaires extrêmement mobiles (plus ou moins empilés) ou des territoires organisés contenant des molécules liant l'ADN... de toute façon il faudra se libérer du modèle héréditaire trop centré sur les unités mutables d'un groupe de liaison que sont les gènes héréditaires (voir cours spécialité). Il faudra abandonner la colinéarité ADN-gènes héréditaires pour sortir de l'impasse gène hériditaire-gène moléculaire. Commencer par le métabolisme de l'ADN pour ne s'attaquer qu'ensuite à son arrangement dans le chromosome... il est possible que le modèle actuel soit assez proche de la réalité mais avec de très nombreux petits ADN... Le tout est d'arriver à proposer un modèle dynamique (qui utilise les fonctions) et non structural (qui repose sur des interactions entre molécules plus ou moins figées dans l'espace). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Le métabolisme des acides
nucléiques et la synthèse
protéique... C'est l'importance relative de ces différents phénomènes qui importe pour avoir une vue d'ensemble. La retrotranscription est-elle un phénomène rare ou bien est-ce le principal mode de fabrication de "gènes" ? Pourquoi n'y a-t-il pas de réplication de l'ARN ? Ce mécanisme n'existe-t-il vraiment pas ou bien ne le connaît-on pas ? |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
En travaux |
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Les "génomes" viraux et des organites ne sont pas des génomes mais des fragments
Schéma le plus
parlant (voir ci-dessus) : |
|
Des génomes procaryotes
: |
|
Des génomes eucaryotes
: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
La séparation ADN génique / ADN extragénique n'est pas la seule pertinente |
|
En effet, la différence entre ADN
génique et ADN extragénique repose sur la
comparaison entre ADN procaryote et eucaryote, en supposant
que la structure est la même. Il y a une autre
manière de voir: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
La structure des gènes procaryotes est peut-être tellement différente de celle des gènes eucaryotes qu'il faut envisager deux modèles différents |
|
Hypothèses classiques venant de la
génétique des procaryotes et étendues
à la génétique des eucaryotes: |
|
Le modèle
instructionniste (un site d'initiation et un
signal de terminaison pour chaque gène, un
système de régulation faisant intervenir
éventuellement des produits d'autres gènes...)
n'est plus le seul envisageable étant donné la
structure du génome de certains eucaryotes.. Ces questions font l'objet du dernier chapitre (en travaux). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Le transfert latéral des gènes... notre génome reflet de notre environnement |
|
Le transfert latéral de gènes (LGT : en anglais Lateral Gene Tranfert) entre espèces est un phénomène dont on commence tout juste à prendre la mesure. En effet, il est de plus en plus fréquent de trouver, dans des écosystèmes comportant plusieurs espèces, des gènes communs qui étaient de prime abord considérés comme spécifiques à l'une ou l'autre des espèces en présence. |
|
Ce phénomène mis en
évidence de façon claire entre populations
bactériennes reste encore mal connu entre eucaryotes.
On sait cependant que les espèces symbiotiques de
bactéries du genre Wolbachia
transfèrent ainsi des segments importants de leur
génome à leur hôte (insecte ou
nématode notamment) -
voir
cours 1ère S. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Des gènes ou des groupes de gènes très répandus * la théorie synthétique de l'évolution a vite fait de parler de conservation de gènes avec un modèle classique de duplication-mutation. Cette vision simpliste doit être dépassée. |
|
Les premiers résultats du développement massif des analyses génomiques automatisées ont tout d'abord surpris par le grand nombre de gènes identiques (ou très voisins) dans les différents phylums*. Lorsque ce sont des groupes entiers de gènes que l'on retrouve, on parle de synténies. Des programmes (voir par exemple le serveur cinteny qui présente quelques exemples de résultats) élaborent des comparaisons phylogénétiques sur des fragments de génomes ou même sur des génomes entiers en utilisant la notion de "reversal distance" (qui est évaluée par le nombre minimum de permutations de segments pour passer d'un génome à un autre). Ces outils mathématiques ont une forte composante théorique et l'on est loin des éventuels mécanismes biologiques. |
|
Si l'on compare les quelques 14.000 gènes codant pour des protéines d'un invertébré (anémone de mer, ver, insecte...) et les 25.000 d'un vertébré, le nombre de gènes plus élevé du second groupe vient principalement de duplications. On cite par exemple les quelques gènes FGF (fibroblast growth factor) d'un invertébré à comparer avec les 20 gènes de type FGF des vertébrés. Les duplications ou répétitions de gènes chez les eucaryotes "évolués" sont une donnée, mais on est loin de savoir intégrer celle-ci aux mécanismes fort mystérieux ayant donné lieu aux différents types d'ADN répété des eucaryotes. |
|
Parmi les étrangetés
"théoriques"...on peut citer: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||