|
ou déficience de l'oxydase de l'acide homogentisique (homogentisate 1,2-dioxygénase ; HGO (HGD est le sigle officiel)) HOMOGENTISIC ACID OXIDASE DEFICIENCY |
||||||
|
Plan |
|
|||||
|
|
||||||
|
Sources: début du travail 10/2002 - mise-en-ligne 12/2007 |
||||||
|
1. L'alcaptonurie est une maladie TRÈS rare |
||||||
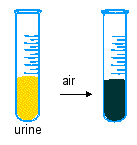 L'urine des malades atteints d'alcaptonurie noircit à l'air... |
L'alcaptonurie est une maladie ancienne (on rapporte à cette maladie des symptômes trouvés sur une momie égyptienne de Harwa datée de 1500 ans avant JC) et surtout très rare (90 cas aux USA, 108 cas en Allemagne, 126 en Tchékoslovaquie, ce qui en fait le pays où l'on a rapporté plus de cas que partout ailleurs). Environ 600 cas dans le monde ont été recensés entre 1956 et 2002. |
Elle est caractérisée par une urine qui noircit à l'air, une pigmentation brun foncé-noire du cartilage et des tissus conjonctifs (ochronosis en anglais - non traduit) et de l'arthrite (inflammation des cartilages articulaires), spécialement au niveau de la colonne vertébrale. Des symptômes secondaires peuvent être associés comme la calcification de valvules aortiques chez les malades ayant dépassé la cinquantaine ou la formations de calculs rénaux ou prostatiques tardifs eux-aussi. Elle est plus fréquente chez les hommes que chez les femmes (environ 75% contre 25% des cas, Garrod 1902). |
||||
|
2 L'histoire d'une
idée à abandonner:
l'alcaptonurie
est une maladie métabolique héréditaire
monogénétique au sens de
Mendel
|
||||||
 Sir Archibald Garrod Un aperçu historique sur le concept "un gène-une enzyme", nettement postérieur et qui ne procède pas du même artifice, est traité sur une page dédiée à Beadle et Tatum |
Les historiens des sciences se sont depuis quelques années attachés à montrer combien les publications d'Archibald Garrod (1857-1936) ont été reprises et diffusées pour en faire des observations fondatrices de l'hérédité mendélienne (voir Gérard Nissim Amzallag, La raison malmenée, 2002, CNRS éditions, p 34s qui cite notamment les travaux de Jan Sapp). |
En affirmant que Garrod exprime, pour la
première fois, en 1902, la possible relation entre
un gène héréditaire et une enzyme
(et donc ce qu'on appellerait maintenant un gène au
sens de la biologie moléculaire c'est à dire
un gène moléculaire)
on le pose en
précurseur génial
méconnu. Les historiens des
sciences parlent du
mythe du "père
fondateur". En tout cas il est clair
que cette date de 1902 est erronée.
|
Revenons donc aux articles originaux et efforçons nous de porter un regard parfois différent de celui que nos maîtres nous ont amené à porter.
|
|||
|
2.1 A. Garrod propose en 1902 une transmission héréditaire mendélienne: un gène autosomal dont la forme récessive correspondrait à l'alcaptonurie, forme métabolique alternative |
||||||
|
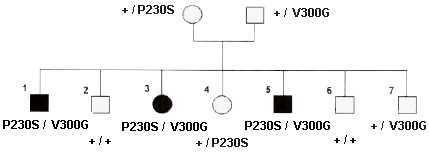
Archibald Garrod a tout d'abord extrait l'homogentisate de l'urine par une méthode plus rapide que ses prédécesseurs (Garrod, 1899, Journal of Physiology, xxiii, 512). Mais il n'a ni découvert cette substance ni proposé en premier de l'associer avec l'alcaptonurie, voir sa revue de la question dans son article de 1902 par exemple (découverte en 1858 puis analysée et extraite en1891). 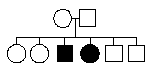 Un mode de transmission autosomal récessif... l'allèle morbide, caché chez les parents (et donc récessif) réapparaît chez les deux enfants malades qui sont alors homozygotes pour cet allèle. Les parents sont forcément hétérozygotes et les enfants sains peuvent être homozygotes sains ou hétérozygotes. |
Garrod était "Physician" (médecin) à l'hôpital des enfants malades de Great Ormondstreet et "Demonstrator of Chemical Pathology" à l'hôpital de St. Bartholomé, lorsqu'il publia en 1902 un article (Garrod AE: The incidence of alkaptonuria: a study in chemical individuality. Lancet II 1902; 1616-20, disponible sur internet en pdf sur le site de l'ESP: http://www.esp.org/ foundations/genetics/ classical/ag-02.pdf ) où il reprend l'interprétation mendélienne de la maladie à partir des propositions de Bateson (W. Bateson. 1902. Mendel's Principles of Heredity, Cambridge). Il se contente de faire une revue des cas connus d'alcaptonurie et d'y ajouter une interprétation autour des liens de consanguinité, notamment des mariages entre cousins germains qui favorisent l'apparition des homozygotes double-récessifs. Étant donné la rareté de la maladie - et donc le non-sens d'une analyse statistique et la difficulté de généraliser les hypothèses - et les informations plus que parcellaires, l'argumentation est plus une voie ouverte qu'une démonstration convaincante. |
|||||
|
Garrod A.E.: The incidence of alkaptonuria: a study in chemical individuality. Lancet II 1902; 1616-20; réedition sur le site de l'ESP, p 11 et 18 du fichier pdf |
« If it be a correct inference from the available facts that the individuals of a species do not conform to an absolutely rigid standard of metabolism, but differ slightly in their chemistry as they do in their structure, it is no more surprising that they should occasionally exhibit conspicuous deviations from the specific type of metabolism than that we should meet with such wide departures from the structural uniformity of the species as the presence of supernumerary digits or transposition of the viscera.» |
|
« Si l'on peut déduire, avec justesse, des faits connus, que les individus d'une espèce ne se conforment pas à une forme standard et rigide de métabolisme, il n'est guère surprenant que des individus puissent présenter des déviations visibles de leur métabolisme spécifique, tout comme l'on pourra trouver de grandes différences au sein de l'uniformité des espèces, comme la présence de doigts surnuméraires ou le déplacement de viscères». |
|||
|
|
||||||
|
2.2 A. Garrod propose entre 1908 et 1923* de relier le gène héréditaire associé à l'alcaptonurie (AKU) à la déficience d'une enzyme spéciale coupant le cycle aromatique de l'acide homogentisique (HGA) |
||||||
|
Inborn
errors of metabolism, A. Garrod,
1909,
les 216 pages de l'édition de 1923 (Second Edition,
London: Henry Frowde and Hodder & Stoughton), sont
accessibles en fac-similé au format pdf sur le site
de l'ESP: http://www.esp.org/books/
|
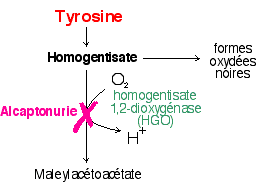
Ce n'est que dans un ouvrage de 1908-1909, Inborn Errors of Metabolism, au chapitre VI puis V (The theory of alcaptonuria), qu'il propose le lien entre l'alcaptonurie (AKU) avec la déficience enzymatique de la dégradation du noyau aromatique de l'acide homogentisique (HGA) pour donner l'acide maléylacétoacétique. L'ochronosis, marquée notamment par le noircissement des cartilages, a été rapportée à l'alcaptonurie depuis 1902 par Albrecht. Elle intervient le plus souvent dans la seconde moitié de la vie des malades. |
Des expériences
sur des malades....
|
||||
|
cliquez
sur l'image pour l'agrandir |
||||||
|
|
||||||
|
Quelques idées nouvelles à partir du travail de N. Amzallag dans la page : De la mutation au problème de la variation. |
|
|||||
|
2.3 Il faut attendre les années 1950-1960 pour que l'enzyme inactive (HGO) soit identifiée chez un patient alcaptonurique |
||||||
|
|
Dans les années 1952-53 des
progrès sont fait dans la connaissance du catabolisme
de la tyrosine chez les mammifères. Mais c'est en
1958 que LaDu et son équipe montrent que le foie d'un
patient atteint d'alcaptonurie présente une
inactivité totale de l'enzyme
homogentisate
1,2-dioxygenase (homogentisate
oxidase)
ou
HGO qui assure la coupure du
cycle aromatique de l'acide homogentisique, alors que toutes
les autres enzymes sont fonctionnelles, montrant ainsi la
grande similitude des systèmes enzymatiques de
dégradation de la tyrosine entre l'homme et les
autres mammifères déjà connus.
|
|||||
|
Un article accessible (en anglais) qui montre les efforts de théorisation sans remettre en cause le modèle moléculaire : |
Genetical
theory and the "inborn errors of
metabolism",
H
Harris, Br Med J.
1970
February 7; 1(5692): 321-27
|
|
||||
|
Pour comparaison voici un article assez complet de 2002 (en anglais aussi mais abondamment illustré), publié lui aussi dans un journal de médecine, et qui, pour le moins qu'on puisse dire, fait allégeance au modèle moléculaire: |
Natural
history of
alkaptonuria,
Phornphutkul C, Introne WJ, Perry MB, Bernardini
I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M,
Anikster Y, Gerber LH, Gahl WA
(2002). N Engl J Med
347:2111-21 Des extraits seront repris plus bas. |
|||||
|
|
||||||
|
Trois exemples de ces
publications (non accessibles
gratuitement) : |
|
|||||
|
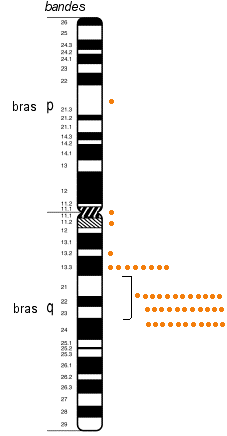
Les résultats présentés ci-contre viennent de la référence 2 ci-dessus: The molecular basis of alkaptonuria, 1996 Pour le vocabulaire utilisé voir le cours de spécialité sur le génie génétique ou technologie de l'ADN recombinant. |
(les positions d'hybridation de la sonde fluorescente (technique de la FISH) de deux clones AKU issus le la base du phage lambda sont notés par des points orange) (les positions discutées "du" gène de l'homogentisate oxidase sont 3q2, 3q13.3-q21, 3q21-q24, 3q21-q23, ou 3q25-q26 ) |
Le gène HGO a été d'abord isolé à partir du champignon Aspergillus nidulans qui possède une enzyme, l'hmgA , équivalente à l'homogentisate oxidase humaine (des gènes d'enzymes à activité équivalente avaient déjà été repérés en 1994 chez la souris et cartographiés sur son chromosome 16). En multipliant et en manipulant ce gène de champignon on a ensuite réussi à isoler par hybridation un fragment d'ADN humain monobrin (ADNc) dans une masse d'ADN extraite d'une cellule humaine et dénaturée. C'est le clonage du gène. |
Cet ADNc de l'HGO humaine est alors utilisé (par technique d'hybridation de type Northern Blot) comme sonde pour trouver l'ARNm dans des extraits de cellules du foie, intestin grêle, colon et prostate. On réalise ainsi des profils d'expression montrant que ce gène est actif (transcrit) dans certains tissus. |
|||
|
|
||||||
|
Carte du gène HGO
(sigle officiel HGD) dans la base de
l'OMIM: séquence de la
protéine HGO (les aa sont notés par des
lettres) |
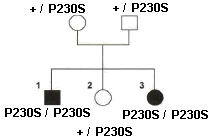
Pour valider le modèle
monogénétique autosomal récessif, deux
familles (14 personnes avec 5 enfants alcaptonuriques) ont
été étudiées pour les mutations
dans le gène HGO
à partir
d'ADN extrait des leucocytes* (et
donc des tissus qui n'expriment pas habituellement l'HGO
alors qu'il aurait été plus logique de le
faire sur des tissus n'exprimant pas l'HGO uniquement chez
les malades). |
famille M
|
||||
|
Pour une réflexion sur les mutations voir De la mutation au problème de la variation |
Le dépistage de "mutations"
(différences mineures de séquence au sein de
la séquence du gène HGO) est relativement
facile car il se fait automatiquement après
séquençage du gène cloné et
amplifié (gène partiel du fait de sa taille
très importante). La difficulté majeure reste la
relation que l'on souhaite établir avec la "mutation"
et l'inactivité d'une protéine. Dans le cas de
HGOP230S
, cette liaison a été établie en
utilisant E. coli comme hôte d'un ADN
étranger portant la "mutation" à analyser.
Pour
HGOV300G
elle est juste supposée. |
familleS
|
||||
|
|
||||||
|
le sigle officiel est désormais HGD et non HGO mais en 2002 les auteurs utilisent encore HGO... |
|
|||||
|
Pour des données plus récentes (voir site de l'NCBI pour le gène HGD). |
Par exemple les auteurs de l'article référencé ci-contre (Ladjouze-Rezig et al., 2006) retrouvent encore l'association de la maladie tant avec les hétérozygotes (ce qui n'est pas compatible avec l'interprétation monogénétique avec une allèle récessif) qu'avec les homozygotes portant des séquences modifiées sans que l'on puisse établir une corrélation entre le génotype et le phénotype. |
|
||||
|
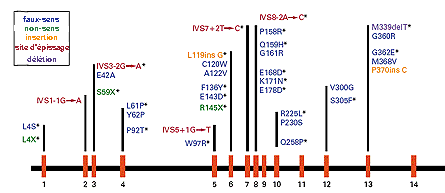
6 ans après l'article historique de 1996 dans Nature Genetics, un article de synthèse, publié dans un journal médical, tentait de faire le point sur les mutations du gène de l'HGO et était arrivé à la même conclusion : |
||||||
|
Natural
history of
alkaptonuria,
Phornphutkul C, Introne WJ, Perry MB, Bernardini
I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M,
Anikster Y, Gerber LH, Gahl WA
(2002). N Engl J Med
347:2111-21 |
Position des mutations des 57 patients/58 cités dans l'article en référence ci-contre dans le gène HGO humain situé sur le chromosome n°3 (bande 3q2). Les bandes verticales oranges correspondent aux exons (régions codantes traduites en chaînes d'aa), les zones noires sont donc des introns (non codantes, excisées lors de la maturation des ARNm ou épissage). Les types de mutations relevées par le séquenceur sont : faux-sens (changement d'aa), non-sens (stop), insertion (ce qui entraîne un décalage de toute la lecture de l'ARNm...), mutation de site d'épissage (splice-site : dans ce type de mutation des nucléotides sont insérés ou délétés juste au niveau des sites d'épissage, ce qui empêche son bon déroulement), délétion (décalage du cadre de lecture). |
L'ADN des patients est obtenu à
partir du sang complet, les exons sont amplifiés par
PCR puis séquencés
automatiquement.
|
||||
|
|
Tous les malades
alcaptonuriques n'ont pas de mutation dans leur gène
HGO
|
Dans cette étude, comme pour la plupart des autres, l'activité enzymatique de l'HGO reste très difficilement accessible et n'a pas été évaluée. On est en droit de penser que le lien, entre mutation et activité locale de la molécule, reste théorique et n'est pas prouvé, expérimentalement, sauf pour quelques cas qui ne permettent pas de généraliser. |
||||
|
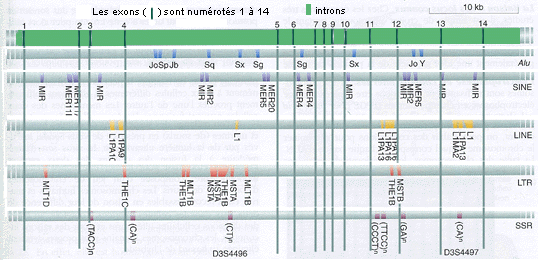
Pour montrer la complexité de ce très grand gène de plus de 50 kb (54.314 bases) voici les marqueurs répétitifs (Alu, SINE (éléments courts), LINE (éléments longs), et transposons LTR et SSR) : certains sont situés dans l'un des 14 exons (en vert plus foncé, les introns étant en vert plus clair et représentant la majeure partie du gène) (in Analyse génétique moderne, De Boeck, 2001, fig.12-14 modifiée) |
|
|||||
|
|
|
|||||
|
Remarque: a et a' désignent deux
allèles du supposé "gène de
l'alcaptonurie" ou "gène de l'HGD" : |
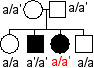 Il existe des malades hétérozygotes ce qui est incompatible avec la récessivité de l'allèle muté a' |
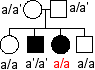 Il existe des individus malades sans mutation dans le gène |
||||
|
|
par exemple, dans les
deux cas présentés ici on peut compliquer le
modèle pour expliquer la transmission
héréditaire: |
|||||
|
Conclusion |
Mais ce qui paraît plus important encore, est le fossé qui se creuse, entre les réseaux métaboliques et les modèles structuraux de fonction locale (fonction enzymatique), plus ou moins rigides, mis en place par différentes techniques de la génétique, et les activités mesurées in vivo, qui semblent reposer sur une individualité et une adaptation qui sortent largement du modèle.
|
|||||
|
3. Des ouvertures pour
comprendre une maladie complexe
|
||||||
|
Au XXIème l'étude
génétique devrait repasser au second plan au
profit d'une vision plus physiologique: la recherche est
toujours ouverte
Pour chaque phénotype (tableau des
symptômes d'un patient) on recherche ce que l'on
appelle génotype et qui est en fait un
séquençage et un typage plus ou moins
précis par marqueurs génétiques. On
s'efforce ensuite d'expliquer la liaison entre ce
"génotype" - plutôt un génotypage
moléculaire - et le "phénotype" - ou
plutôt les caractéristiques physiologiques et
biochimiques individuelles- d'un malade. On fait appel la
plupart du temps à des "gènes modulateurs"
hypothétiques pour expliquer des "phénotypes"
surprenants. Pour ce qui est de l'identification du
gène héréditaire et du gène
moléculaire, la question reste ouverte.
L'interprétation héréditaire est
cohérente et se suffit à elle-même. Pour
l'instant il n'existe pas d'autre théorie
héréditaire à ma connaissance. |
||||||
|
A la recherche des populations cellulaires impliquées... Je sais qu'il est surprenant de proposer d'affirmer que l'alcaptonurie est d'abord une maladie de l'enfance car de nombreux symptômes apparaissent tardivement, surtout au niveau musculo-squelettique ou encore de la peau. Nombre de personnes atteintes d'alcaptonurie sont asymptomatiques avant l'âge de 20-30 ans. Ces symptômes plus ou moins tardifs sont reliés à l'accumulation d'acide homogentisique (AHG) et de son produit d'oxydation l'acide benzoquinone acétique (ABQA). Mais si l'on s'efforce de comprendre comment les différents symptômes sont reliés entre eux, on peut penser que l'alcaptonurie touche des populations cellulaires qui ne sont pas sans lien au cours du développement: les cellules du foie (hépatocytes - en sachant que c'est le foie qui est le lieu de l'hématopoièse chez le fœtus), les cellules de la peau (mélanocytes, qui comprennent des cellules claires ("de Masson") et des cellules dendritiques qui dériveraient de mélanoblastes embryonnaires venant des crêtes neurales de l'embryon) et les articulations sans que l'on sache précisément quelle population cellulaire est touchée (mais étant donné d'une part les contraintes importantes auxquelles sont soumises les articulations et d'autre part l'importance du système vasculaire et immunitaire au niveau de celles-ci, il est probable que ce soient encore des populations responsables de l'immunité locale - comme des cellules dendritiques - qui soient ici touchées). On pourrait donc penser que ces populations cellulaires, fortement impliquées dans la fonction immunitaire, seraient les témoins de la maladie précoce. Par contre les travaux de biologie moléculaire qui ont cherché à localiser les enzymes HGO déficientes ont trouvé des profils d'expression au niveau de foie, du rein, de l'intestin grêle et de la prostate. Ces tissus ne semblent pourtant pas être impliqués directement dans les symptômes invalidants. Il y a donc en quelque-sorte un fossé, entre le déterminisme métabolique proposé et les observations cliniques, qui mérite d'être souligné. Une maladie du collagène |
||||||
|
L'idée que l'alcaptonurie puisse venir d'un anomalie de développement dans des populations cellulaires synthétisant du collagène est assez naturelle. |
L'alcaptonurie est placée dans les maladies du collagène (pour des données sur cette molécule, voir cours de 1èreS). En effet, causant des troubles articulaires à partir d'un certain âge, l'alcaptonurie serait une maladie du collagène dans la mesure où l'HGA bloquerait une enzyme, la lysyl hydroxylase, fragilisant la molécule de collagène devenue ainsi hydroxylysine-déficiente. Mais il serait tout aussi cohérent de penser que les dérèglements sont bien antérieurs et se placent lors de la période embryonnaire. |
Diseases
of the collagen molecule, C. I. Levene, J Clin
Pathol Suppl (R Coll Pathol). 1978; 12:
82-94, |
Comme les cellules de la peau ont été prises comme exemple dans le cours de première S, il me paraît judicieux de faire des rapprochements entre les connaissances acquises en 1ère et les données de cette page. |
|||
|
Une maladie pigmentaire / une maladie du catabolisme |
||||||
|
Les populations de cellules touchées par les formes adultes de la maladie sont celles de la peau et des cartilages. On peut donc essayer de faire un lien entre les phénotypes cliniques des malades et les fonctions de ces populations cellulaires. |
Il existe tout d'abord un lien entre les produits du catabolisme (et notamment du catabolisme peptidique) et les pigmentations. Si les fonctions des pigmentations sont nombreuses on cite le plus souvent, d'une part la protection (des rayonnements solaires, camouflage...), et d'autre part le stockage des déchets; la pigmentation devient ainsi une forme d'excrétion de substances toxiques. |
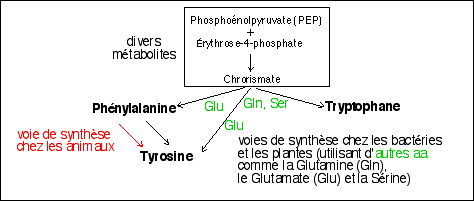
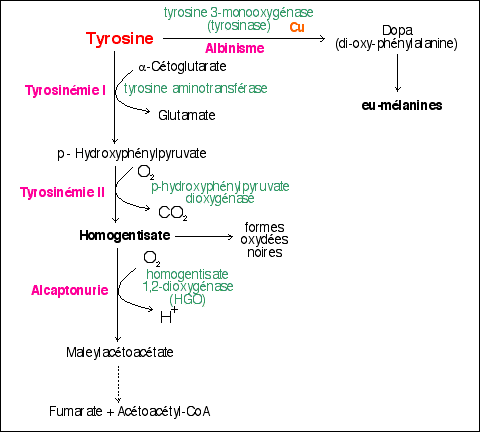
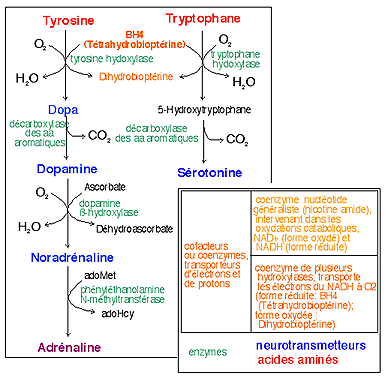
Mais il existe d'autres voies formant un réseau. Voici un aperçu de quelques autres maladies métaboliques qui font intervenir la tyrosine. Même si le modèle de leur transmission héréditaire est parfois contesté et considéré comme complexe maintenant, elles sont toutes été, historiquement, rapportées à la transmission d'un gène autosomique, la maladie correspondant à un allèle récessif. |
Les Tyrosinémies I et II sont des maladies métaboliques qui ont été associées à des déficits enzymatiques intervenant dans le catabolisme de la tyrosine. Une certaine forme d'albinisme (décoloration pigmentaire) peut aussi être reliée à la déficience dans la synthèse de la Dopa à partir de la tyrosine (voir réactions). |
|||
|
4
maladies métaboliques à
forte composante
héréditaire
liées au catabolisme de la
tyrosine
(d'après Lehninger, p 529,
fig 17-26)
|
|
|||||
|
|
||||||
{kind=link}