|
|
|
ou l'idiotie phénylpyruvique |
|
mise-à-jour 12/2007 |
||
|
|
||||||
|
retour page d'accueil, plan du cours de 1èreS, panorama |
|
Le but de cette page n'est pas une étude médicale ni encore moins scientifique exhaustive de cette maladie, qui sort de mon champ de compétence (professeur de SVT) mais un recadrage des notions au programme (génotype, phénotype, liaison avec l'environnement) d'une part avec les données de vulgarisation scientifique facilement accessibles (et parfois des articles à accès réservé), et d'autre part avec une biologie théorique plus ouverte que la seule biologie moléculaire. Je ne prétends pas mieux comprendre que les autres personnes cette maladie mais j'espère que cette page aidera un élève à faire le tri entre ce qui est connaissance scientifique assurée, magie explicative de certains concepts et espoir teinté d'idéologie. |
||||
|
|
|
|
||||
|
Plan |
|
|||||
|
lien avec une page historique sur l'alcaptonurie |
|
|||||
|
1. Données
préliminaires physico-biochimiques sur les aa
aromatiques
|
||||||
|
|
|
|||||
|
|
|
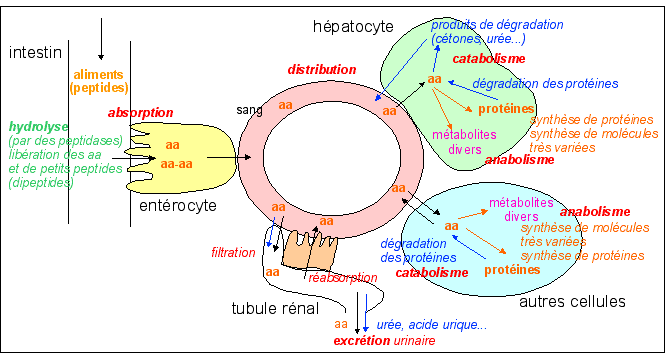
Les acides aminés (aa) sont les constituants des protéines mais ont de nombreux autres rôles dans les cellules car ils sont le point de départ de la synthèse de nombreuses molécules originales (métabolites). Les aa sont trouvés dans l'alimentation mais certains peuvent aussi être synthétisés. L'organisme ne peut pas les stocker; leur vitesse et taux de renouvellement dépend des cellules et surtout de l'âge de l'organisme (et donc de l'importance des synthèses de protéines). |
|
Remarque: |
||
|
|
Quelques grandes lignes du métabolisme des acides aminés (aa): ils sont absorbés, distribués dégradés et excrétés. Le métabolisme comprend l'ensemble des réactions chimiques et peut se répartir en anabolisme (synthèse) et catabolisme (dégradation). La synthèse des protéines à partir d'aa fait partie de l'anabolisme, même si les aa sont utilisés, tout comme la synthèse de la tyrosine à partir de la phénylalanine. De même, la dégradation des protéines fait partie du catabolisme, même si elle libère des aa. |
|||||
|
|
|
|
||||
|
|
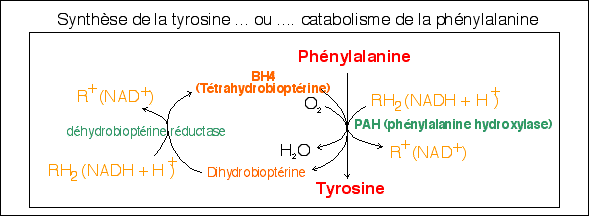
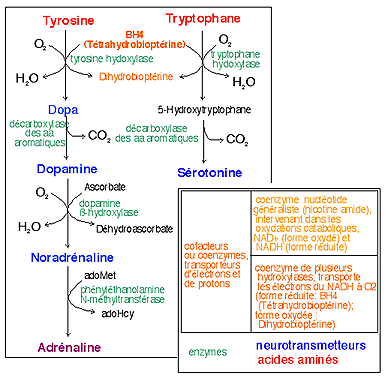
Deux acides aminés possèdent un noyau aromatique (à 6 carbones reliés cycliquement par des liaisons covalentes dont certains électrons sont mis en commun (électrons º); le cycle est représenté avec 3 doubles liaisons et 3 liaisons simple lorsqu'il y a des groupements supplémentaires liés aux carbones ou avec un cercle central si le noyau benzène est intact): la phénylalanine (Phe) et la tyrosine (Tyr). Ces deux acides aminés sont à l'origine de la synthèse de nombreuses molécules à cycle aromatique qui interviennent dans le métabolisme: des hormones et autres messagers (catécholamines (Dopa, dopamine, noradrénaline, adrénaline), sérotonine, hormones thyroïdiennes), pigment (mélanine) (voir quelques réactions). |
|
La dégradation de ces acides aminés donnent des corps cétoniques. Chez les animaux normalement seule la phénylalanine est un acide aminé indispensable (qui doit être trouvé dans l'alimentation); la tyrosine est normalement synthétisée par l'organisme à partir de la phénylalanine (voir schéma ci-dessous, explication des couleurs sur le schéma). |
|||
|
|
|
|
||||
|
|
|
|
||||
|
Une réaction chimique peut avoir des significations différentes selon le contexte cellulaire |
|
Il est important de comprendre que les réactions métaboliques sont extrêmement imbriquées et que selon les cellules et, pour les mêmes cellules, selon leur âge, ou encore, selon l'état physiologique d'un individu, une réaction chimique peut ne pas avoir la même signification. |
|
* La conversion de la phénylalanine en tyrosine peut être tout d'abord une voie de synthèse de la tyrosine - anabolisme - (par exemple dans un mélanocyte de la peau qui synthétise la mélanine à partir de la tyrosine). Une telle cellule consomme une certaine quantité de phénylalanine qu'elle doit absorber à partir des aa sanguins circulants (parce que l'organisme ne stocke pas les aa). |
|
* La conversion de la phénylalanine en tyrosine peut ensuite être une voie de dégradation de la phénylalanine - catabolisme - par exemple dans les cellules hépatiques qui dégradent les acides aminés en corps cétoniques et en urée. |
|
|
|
|
||||
|
Les réactions métaboliques in vivo forment un réseau stable (la stabilité c'est l'homéostase qui résulte de la fonction) |
|
En biologie moléculaire, un gène moléculaire spécifique d'une enzyme (comme la PAH ci-dessus) est bien évidemment NÉCESSAIRE à la synthèse de cette enzyme mais il n'est PAS SUFFISANT pour que l'enzyme soit fonctionnelle. |
|
La fonction enzymatique n'est pas comprise uniquement au niveau moléculaire (ce que l'on peut appeler sa fonction locale que l'on peut explorer chimiquement (voir cours de premièreS sur les enzymes) mais au niveau de sa fonction cellulaire ou globale, qui ne peut être explorée aussi facilement. La PAH par exemple ne peut pas être considérée comme fonctionnelle tant que son niveau de concentration, son emplacement (dans le cytoplasme, en complexe, ou à la surface de membranes), sa vitesse de renouvellement, ... , sont suffisant dans cette cellule. Cet ensemble de fonctions (ou cette fonction globale) ne peut résulter que d'un fonctionnement intégré de la cellule qui ne peut pas être décrite de façon exhaustive par la somme d'interactions moléculaires, dans l'état actuel de nos connaissances. Pour comprendre ce fonctionnement cellulaire global il faut faire appel à des fonctions globales comme le travail de relation, de nutrition ou de reproduction (voir Qu'est-ce que la vie ?, cours de 1èreS sur le paysage phénotypique). |
||
|
|
|
|
||||
|
Un même gène moléculaire unique peut être utilisé pour synthétiser des enzymes différentes |
|
Par exemple la tyrosinase (ou thyrosine 3-monooxygénase) est une enzyme (à Cu2+) isolée à partir des mélanocytes qui transforme la tyrosine en Dopa; On l'a assimilé à la tyrosine hydroxylase (TH) qui catalyse la même réaction dans les neurones catécholaminergiques avec le BH4 comme cofacteur (voir réactions) . |
|
En fait, et du fait du rôle important dévolu à la TH dans les aliénations mentales comme le complexe maniaco-dépressif, les recherches on été intenses (voir site en anglais pour des détails OMIM=http://www.ncbi. nlm.nih.gov /entrez/dispomim.cgi ?id=191290). On a ainsi isolé, depuis les années 1985, un unique gène (assez voisin de celui de la PAH) qui a une taille d'environ 8 kb et qui serait transcrit en un unique et immense ARNm. Cet ARNm subirait ensuite différentes maturations (on parle d'épissage alternatif) selon les cellules où il serait produit. L'enzyme TH aurait donc 3 ou 4 formes différentes (avec des segments manquants ou supplémentaires de 4 et 27 aa). |
|
On connaît des cas d'épissage alternatif qui conduisent à des protéines aux rôles très différents (le gène moléculaire de la calcitonine par exemple qui peut aussi être utilisé pour synthétiser un neurotransmetteur). |
|
2. L'alcaptonurie ne doit
plus être enseignée comme exemple d'une maladie
à mode de transmission monogénétique
autosomal récessif
|
||||||
|
voir page historique sur l' alcaptonurie |
|
L'alcaptonurie (AKU) est la première des maladies métaboliques à avoir été associée par A. Garrod à une transmission héréditaire monogénétique mendélienne (1902) puis à des mutations dans un gène unique (associé à l'enzyme HGD: homogentisate 1,2-dioxygenase ou homogentisate oxidase, HGO a été employé comme sigle par le passé). |
||||
|
|
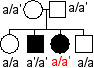
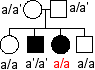
Illustration ci-contre des incohérences auxquelles les chercheurs sont confrontés: |
|
a et a' désignent deux
allèles du supposé "gène de
l'alcaptonurie" ou "gène de l'HGD" : |
|||
|
|
Il existe des malades hétérozygotes ce qui est incompatible avec la récessivité de l'allèle muté a' (cas 1) Il existe des individus malades sans mutation dans le gène (cas 2) |
|
|
|
|
|
|
|
|
|||||
|
|
||||||
|
3. La
phénylcétonurie: des symptômes, des
malades, une maladie ?
|
||||||
|
l'idée d'un service éducatif national de traduction scientifique me paraît à creuser... |
|
Sources: |
||||
|
La phénylcétonurie
est une question clé ACTUELLE, un "laboratoire"
où s'affrontent les points de vue les plus novateurs
et les efforts intéressés (l'aspect financier
n'est pas négligeable) d'équipes en vue. Je ne
prétends pas ici avoir réussi à cerner
le problème. J'espère simplement que les
questions que je pose sont de bonnes
questions.
|
Je ne crois pas que les élèves puissent suivre facilement le débat (pour eux, je renvoie à l'alcaptonurie qui est un modèle plus simple et surtout beaucoup moins étudié car il n'y a pas d'enjeux financiers du fait de la rareté de la maladie); ce travail est donc plutôt destiné aux collègues ou aux élèves du supérieur qui ne seraient pas encore trop impliqués dans une modèle particulier. |
|||||
|
|
|
|||||
|
|
|
|
|
Des malades dont le
point commun est de présenter un retard
mental avec une odeur
caractéristique des urines.
|
||
|
|
Si la publication originale de A Fölling est introuvable (Fölling A. Excretion of phenylpyruvic acid in urine as a metabolic anomaly in connection with imbecility. Nord Med Tidskr. 1934;8: 1054-105), on peut néanmoins trouver un article d'histoire des sciences en libre accès ci-contre (<----): |
|
Où l'on apprend notamment |
|||
|
Pour des données médicales simples, se référer à l'article sur Orphanet. |
|
l'odeur et le
phénylpyruvate
|
|
|
||
|
|
|
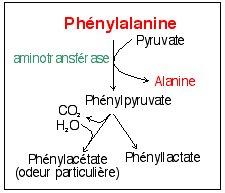
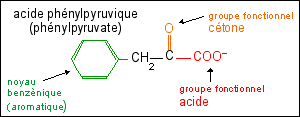
L'acide phénylpyruvique (ou
phénylpyruvate) - qui a aussi un groupement
fonctionnel cétone, d'où le nom de
phénylcétone
- est relié au catabolisme de la Phe
(voir
ci-contre) mais ce n'est pas lui qui
confère directement l'odeur particulière aux
urines mais un de ses produits de dégradation
l'acide phénylacétique. |
|
D'où vient l'acide
phénylpyruvique? La Phe sanguine passe à
travers le filtre rénal et est normalement
réabsorbée totalement
(voir
généralités sur les aa dans la
partie
1). C'est dans le foie que la Phe
est transformée en phénylpyruvate et dans
d'autres métabolites. Le symptôme urinaire est donc sans aucun doute à relier à une cause hépatique, pouvant être multiple. On trouve encore le terme d''oligophrénie phénylpyruvique pour désigner la PKU (du grec, oligos = peu, phrénos = relatif à l'esprit - un dictionnaire de terminologie médicale bien utile, avec racines : BioTop); équivalent donc d'idiotie phénylpyruvique.
|
||
|
|
|
les
hyperphénylalaninémies
|
|
|
||
|
|
|
Il faut bien noter qu'il n'y a pas de Phe
dans l'urine (ce qui correspondrait à une
phénylalaninurie). Les diagnostics de PKU associent donc la phénylalaninémie à d'autres facteurs dont la phénylcétonurie (et la présence d'autres métabolites de la Phe dans les urines).
|
|
Que signifient des taux sanguins de Phe
trop élevés par rapport à une norme
établie à partir de très nombreuses
mesures ? Nous sommes clairement dans une étude
physiologique et épidémiologique. Une réponse simple à cette question n'est donc pas accessible. |
|
Les causes pouvant affecter les éléments du réseau métabolique où interviennent la Phe et la Tyr sont tellement nombreuses qu'il n'est pas concevable de considérer que l'on puisse en dresser un tableau compréhensif synthéthique. Toute explication procède nécessairement d'une vue simplifiée. On s'est donc focalisé sur les métabolismes qui semblent affectés chez les malades. C'est pour cela que la vision molécularo-génétique s'est imposée comme une vision claire... qui s'est complexifiée au point de devenir obscure. On ne peut pas revenir à un réseau métabolique sans tenir compte de tous les résultats épidémiologiques qui se sont accumulés dans le domaine de la génétique de la PKU. |
|
|
|
le retard mental
(encéphalopathie)
|
|
|
||
|
|
|
Plus qu'un retard, il s'agit d'un
arrêt de développement mental vers l'âge
de 3-5 ans en cas d'absence de traitement. Il y a une corrélation entre la concentration de Phe plasmatique et la détérioration neurologique, les anomalies électroencéphalographiques (d'EEG) et les taux sanguins de neurotransmetteurs, en particulier la dopamine. |
|
|
||
|
|
|
|
|
|
||
|
|
|
3.2
L'interprétation la plus conventionnelle, on ne sort
pas du paradigme génétique, on rajoute une
couche d'épigénétique et on invente des
gènes modificateurs
|
||||
|
un article de revue
récent
en
bleu
les commentaires personnels, en
rouge
des parties qui me paraissent
importantes |
|
The
PAH gene, phenylketonuria, and a paradigm shift,
Charles R. Scriver, 2007, Human Mutation, Volume 28,
Issue 9 , Pages 831 - 845 |
||||
|
|
|
|
|
|||
|
|
|
|
||||
|
|
|
|
|
Misfolded Proteins and
AllelicVariation |
|
Protéines
mal-repliées et variation allélique |
|
|
|
|
|
|
||
|
|
|
|
|
les gènes
modificateurs
un gène modificateur = un gène qui affecte l'expression phénotypique d'un autre gène
|
|
Les "gènes modificateurs" ou encore "gènes modulateurs" (en anglais "modifier gene" ou "modifier locus", "modificator gene"..) sont des inventions directement issues du paradigme génétique dans lequel se sont enfermés les molécularistes. Plutôt que d'abandonner la liaison moribonde entre le génotype et le phénotype dans une vision héréditaire mendélienne on se raccroche aux branches en inventant le gène à tout faire. Ce serait pathétique, si ce terme n'était pas employé dans des publications scientifiques. Cette invention a tout simplement un goût de mystification. |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
À la recherche
d'exemples pour revenir à une vision plus
intégrée...
|
||
|
Il n'y a pas de consensus suffisant autour d'une autre théorie. Toute tentative se fait forcément à contre-courant et nécessite donc une bonne dose de prudence si l'auteur ne veut pas se mettre à l'écart de la société scientifique. |
|
Dans le cas de la glycémie on a forgé nos anciens modèles à partir de l'idée d'une constance - de la glycémie- à l'image de l'homéostasie bernardienne. J'ai expliqué par ailleurs combien ces modèles cybernétiques me paraissent inadaptés pour un organisme vivant dont l'homéostase est celle des fonctions et non des paramètres (voir glycémie). |
|
Dans le cas des acides aminés, et tout particulièrement des acides aminés aromatiques, qui interviennent dans de très nombreux systèmes, et n'ayant pu s'appuyer sur la constance de paramètres mesurables, on s'est efforcé de présenter l'homéostasie comme résultant d'une constante de l'activité enzymatique de quelques enzymes clés: ici la PAH et le cofacteur BH4. |
|
Dans l'article de C.R. Scriver étudié ci-dessus on trouve un extrait d'un travail qu'il a réalisé autour du concept d'homéostase dans le cadre de la PKU. je vais partir de cette idée pour essayer de suivre d'autres voies. Remarque: |
|
|
|
|
|
|
||
|
Comment changer cette représentation en paysage épigénétique ?
|
|
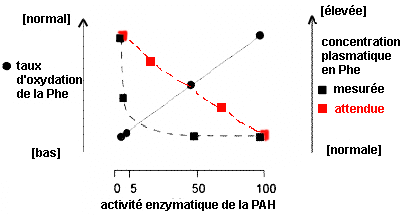
Si l'on considère la réaction la plus connue, l'oxydation de la Phe en Tyr par la PAH (voir représentation ci-dessus) est une réaction enzymatique sous la dépendance de la concentration en Phe, et de l'activité de la PAH, en supposant le cofacteur BH4 présent et actif et le dioxygène accessible. On s'attend donc à obtenir
théoriquement une courbe d'activité qui aurait
l'allure approximative de la courbe en rouge que j'ai
ajouté à la figure originale
(ci-contre à
gauche).
Pour un aperçu des modèles catastrophiques voir page sur La théorie des modèles de René Thom et une page complémentaire (attention applet Java à télécharger 1Mo). |
||||
|
légende originale |
|
|
|
|||
|
|
|
|
|
|||
|
En travaux |
|
|
|
|
||