La phénylcétonurie
(PKU, PCU),
ou
l'idiotie
phénylpyruvique,
Cette ancienne page (que je
laisse comme archive) a été remplacée en
décembre 2007 par une nouvelle
page
Sources:
http://www.inrp.fr/Acces/biotic/gpe/dossiers/phenylcetonurie/accueil.htm;
cette riche source fournit
des outils pour modéliser des enzymes (PAH) et cofacteurs
(BH4) (que peut-on en faire dans une classe de terminale lambda ?)
mais n'approfondit pas la liaison génotype-phénotype,
notamment le rôle de l'environnement sur les gènes (ce
qui est bien la question soulevée par le programme. Le terme
de phénotype y est pris aussi bien au niveau clinique (ce qui
n'a aucun sens à mon avis, il s'agit de symptômes d'une
maladie métabolique, génétique, voir page sur
le
phénotype)
qu'au niveau cellulaire (appelé à tort
moléculaire). Les cellules hépatiques qui
possèdent tel ou tel allèle du gène de la PAH
semblent exprimer toujours (?) les deux allèles, sans que les
conditions de cette expression, soient vraiment
abordées...
Principes de biochimie, Lehninger, Nelson et Cox, 1994,
Médecine-Sciences-Flammarion, p 525s,
714s, pour tout ce qui concerne le
catabolisme des acides aminés et la synthèse des
neuroamines.
Pour l'aspect médical:
http://www.med.univ-rennes1.fr/etud/pediatrie/erreurs-metabolisme.htm
http://www.med.univ-rennes1.fr/etud/pediatrie/phenylcetonurie.htm
retour page d'accueil, plan
du cours de 1èreS, cours de
1èreS
Le but de cette page n'est pas une
étude médicale ni encore moins scientifique exhaustive
de cette maladie, qui sort de mon champ de compétence
(professeur de SVT) mais un recadrage des notions au programme
(génotype, phénotype, liaison avec l'environnement)
avec les données de vulgarisation scientifique facilement
accessibles pour cet exemple.
Cet exemple présente un intérêt
historique car son étude intervient dans le prolongement de la
proposition d'Archibald Garrod en 1909 (voir histoire
de la génétique) au sujet de la liaison entre
l'alcaptonurie (maladie métabolique sans autre
effet que la présence de pigment noirs dans les urines et
l'apparition d'arthrites tardives) et le déficit
métabolique en une unique enzyme
(l'homogentisate 1,2-déshydrogénase) intervenant
dans le catabolisme de la tyrosine. Cette hypothèse fut
extraordinairement féconde puisque l'on connaît
déjà une dizaine de maladies métabolique du
catabolisme des acides aminés que l'on a parfois
accompagné d'une étude génétique des
malades.
En effet la phénylcétonurie, maladie dont le
symptôme le plus grave est une encéphalopathie (maladie
de l'encéphale, retard mental et parfois moteur), induit aussi
une odeur caractéristique des urines expliquée pour la
première fois par Fölling comme étant due à
la présence d'acide phényl-pyruvique relié au
catabolisme de la phénylalanine. La présence de
phénylalanine en excès dans le sang
(hyperphénylalaninémie) est tout aussi
caractéristique que la présence d'acide
phénylpyruvique dans les urines (d'où le nom de
phénylcétonurie). Dès 1950, Bickel,
proposa de soumettre les enfants atteints de cette maladie à
un régime dépourvu ou très pauvre en
phénylalanine ce qui permettait d'éviter
l'encéphalopathie.
Des symptômes, des malades, une
maladie
Pour l'aspect médical, voir les sites ci-dessus. Tous les
malades ne présentent pas des symptômes identiques et
pour un symptôme identique, les causes identifiées
peuvent parfois être différentes chez deux malades.
L'important est de comprendre que le taux anormalement
élevé de phénylalanine dans le sang
(hyperphénylalaninémie) est dangereux essentiellement
lors du développement des cellules nerveuses,
c'est-à-dire chez le jeune enfant (avant 10 ans) et chez
l'embryon et le fœtus étant donné que l'acide
aminé, de petite taille passe la barrière placentaire
et va du sang de la mère à celui de l'enfant et
vis-versa.
Une maladie métabolique
On distingue maintenant au moins trois types de déficits
métaboliques chez les malades:
- soit une déficience de l'enzyme phénylalanine
hydroxylase (PAH), plus ou moins poussée
(depuis une activité nulle,forme classique
P.C.U.,à une activité à 3 % pour la forme
atypique ou "méditerranéenne", en passant par une
activité de 3 à 6 %, pour la forme
modérée),
- soit une déficience du cofacteur intervenant conjointement
avec cette enzyme: la tétrahydrobioptérine (BH4),
- soit encore une déficience de l'enzyme qui restaure ce
cofacteur : la dihydrobioptérine réductase (connue
aussi pour le métabolisme de la dopamine et de la
sérotonine, autres neurotransmetteurs, ce qui explique des
déficiences nerveuses plus étendues dans ce cas). Le
problème étant que le métabolisme est
fondamentalement intégré, et donc forme un
réseau interconnecté de réactions chimiques, ces
enzymes et cofacteurs interviennent dans des réactions
différentes selon les cellules et selon les organismes, ce qui
conduit à des conséquences complexes à
identifier. Ce ne peut donc être le but de l'étude de
cet exemple en classe de terminale.
Une étude génétique des malades et de leur
famille
De très nombreux allèles ont été
séquencés pour la PAH et la BH4 (voir site de l'inrp).
Chaque forme plus ou moins efficace d'une enzyme ou du cofacteur
correspond à un allèle dont on connaît la
séquence. Le phénotype (forme
d'un allèle dans une cellule) associé
à un génotype (ensemble des
allèles d'un gène dans une cellule)
donné n'est par contre pas évident à
présenter.
Du point de vue THÉORIQUE:
* chaque cellule hépatique ou nerveuse,
diploïde, possède deux allèles de la
PAH (un venant de la cellule sexuelle paternelle, l'autre de la
cellule sexuelle maternelle)
* les deux allèles sont exprimés avec la même
intensité dans chaque cellule; ce qui fait que si une cellule
est homozygote pour le gène de la PAH avec
l'allèle correspondant à la forme la plus efficace et
courante de l'enzyme (forme saine notée par exemple PAH+),
elle produira une quantité Q d'enzyme PAH saine par
unité de temps. Dans les mêmes conditions
métaboliques, une cellule hépatique
hétérozygote pour ce gène avec par
exemple un allèle sain (PAH+) et un allèle
correspondant à la forme enzymatique présentant une
activité enzymatique nulle (notée PAH0 par exemple),
produira donc une quantité d'enzyme PAH égale à
Q/2.
Dans la réalité:
Les sources ne mentionnent que peu de cas d'activité
enzymatique modulée par l'environnement (différence
entre l'expression des gènes dans une cellule hépatique
jeune et vieille ou entre une cellule hépatique et une cellule
nerveuse...) ou, plus probable encore, une différence
d'activité de transcription (ADN-->ARNm) des gènes
de la PAH modulée en fonction de l'allèle et surtout de
l'activité des enzymes produites (rétrocontrôle
sur les gènes). Mais, plus gênant encore, ces sources
mélangent le tableau clinique (les symptômes) et les
phénotypes cellulaires (qui ne sont pas décrits).
Le site de l'INRP (http://www.inrp.fr/Acces/biotic/gpe/dossiers/phenylcetonurie/html/modulateurs.htm)
propose le cas de deux frères présentant une
concentration en phénylalanine du sang supérieure
à 25 mg.dL-1 (le taux courant est
inférieur à 4 mg/dL-1 et l'acide
phénylpyruvique n'est retrouvé dans les urines
qu'à partir de 15 à 20 mg/dL-1 ). Le
premier, âgé de 10 ans, avait un développement
neurologique normal, alors que le second, âgé de 6 ans,
présentait un retard mental sévère. L'analyse
génétique pratiquée a
révélé que tous les deux avaient le même
génotype (homozygote ?),
avec (pour chaque allèle
?) les mutations Ser349Pro et une délétion
d'un nucléotide en position 55 entraînant un codant stop
anticipé et donc une PAH très peu efficace et une
hyperphénylalaninémie très forte.
Cette différence clinique peut avoir plusieurs
interprétations dans le cadre de notre vision ouverte qui se
réfère aux trois niveaux d'information du vivant
(génétique, cytoplasmique et extracellulaire ou
environnemental):
- une cause strictement génétique (on peut
penser par exemple à l'existence de gènes
modulateurs intervenant directement sur le gène de la PAH
dans les cellules où ce gène est déficient);
- une cause cytoplasmique au niveau des cellules dont le
gène de la PAH est déficient qui puisse compenser,
métaboliquement par exemple l'absence de catabolisme de la
phénylalanine (voie alternative, transport des
métabolites...); chaque cellule modulant son
phénotype à partir de son génotype; c'est ce
que l'on appelle la norme de réaction (d'un
génotype) qui est la somme des phénotypes possibles
pour une cellule donnée d'un certain génotype en
fonction des environnements dans lesquels elle se trouve.
- une cause extracellulaire (environnementale) physiologique
qui se situe en dehors des cellules où le gène de la
PAH est déficient. Bien évidemment, ce sont encore des
cellules qui agissent même si on considère les
mécanismes à un niveau d'intégration (hormonal,
nerveux...) qui ne se réduit pas à un aspect
moléculaire; cette hypothèse est aussi proposée
par les auteurs du site de l'INRP qui font état de recherches
concernant des canaux spécifiques qui font entrer les acides
aminés (comme la phénylalanine) dans les cellules
nerveuses: on imagine aisément que si ces canaux sont moins
actifs dans certaines lignées cellulaires, ils peuvent
permettre de compenser les effets dangereux d'une
hyperphénylalaninémie.
Ces trois niveaux, théoriques, sont bien
évidemment difficiles à délimiter. Ce qui est
rassurant car il ne s'agit pas de les opposer. En effet, ce ne sont
que des outils qui permettent de cerner une question.
Remarque:
les auteurs du site de l'INRP, comme de nombreux auteurs de manuels
scolaires, présentent les régimes alimentaires suivis
par certains malades comme des facteurs environnementaux susceptibles
de modifier l'expression des génotypes des cellules
hépatiques de ces personnes. Cette vision est surprenante car
il ne s'agit pas d'un contrôle de l'environnement (l'individu
étant considéré comme une société
de cellules en évolution) mais d'un comportement qui
relève à mon sens de la médecine (art de
soigner). Certes, le comportement de l'homme est un
élément de sa biologie, mais je pense, encore une fois,
qu'il est vraiment néfaste de vouloir utiliser sans
précautions le terme de phénotype au-delà de la
cellule.
Une maladie héréditaire
Du point de vue héréditaire, si l'on associe de
façon absolue la maladie à la présence de deux
allèles délétères de la PAH dans les
cellules hépatiques par exemple, on a donc un seul
gène (maladie dite monogénétique) qui, selon
les lois de la génétique mendélienne et
morganienne (voir cours de spécialité
de terminale ou l'histoire
de la génétique) se
transmet avec un chromosome (le chromosome 12, un autosome,
c'est-à-dire un chromosome non sexuel). Étant
donné qu'une cellule qui ne possède qu'un allèle
morbide (PAH déficiente par exemple) présente une
activité presque normale de la PAH, on dit que l'allèle
morbide est donc récessif par rapport à
l'allèle sain, qui est dit dominant. Étant
donné la variation quasi continue entre une activité
nulle de la PAH et une activité maximale, selon le
polymorphisme allèlique (nombreuses mutations
conduisant à de nombreux allèles, voir
le site de l'INRP: http://www.inrp.fr/Acces/biotic/gpe/dossiers/phenylcetonurie/html/mutations.htm),
cette distinction est tout à fait simpliste. Une chose est
sûre, c'est qu'un enfant hérite bien des allèles
maternels et paternels, sauf anomalie et donc que son génotype
est bien fixé par l'hérédité
mendélienne. Mais ce qui est moins déterminé,
c'est l'utilisation que ces cellules vont faire de ce
génotype, et donc quel sera son phénotype et partant,
si il sera malade ou non. Le génotype est bien une
donnée mais il n'y a pas de causalité directe entre le
phénotype et le génotype. Avec les mots d'un
embryologiste: «Dans tous les cas, l'organisme hérite
de l'aptitude à répondre aux signaux de
l'environnement, mais il n'y a pas de prédiction du
phénotype par le génotype.»
(Biologie du développement, Scott F.
Gilbert, De Boeck Université, 1996, p 74).
Donc, cette vision simpliste issue de l'analyse
héréditaire classique doit faire place à une
nouvelle hérédité basée sur l'analyse des
allèles et des phénotypes cellulaires, au sein de
organes appartenant à des individus d'une même fratrie.
Analyse qui pourrait révéler encore bien des surprises
sur les "lois de l'hérédité", non pas
génétique mais cellulaire.
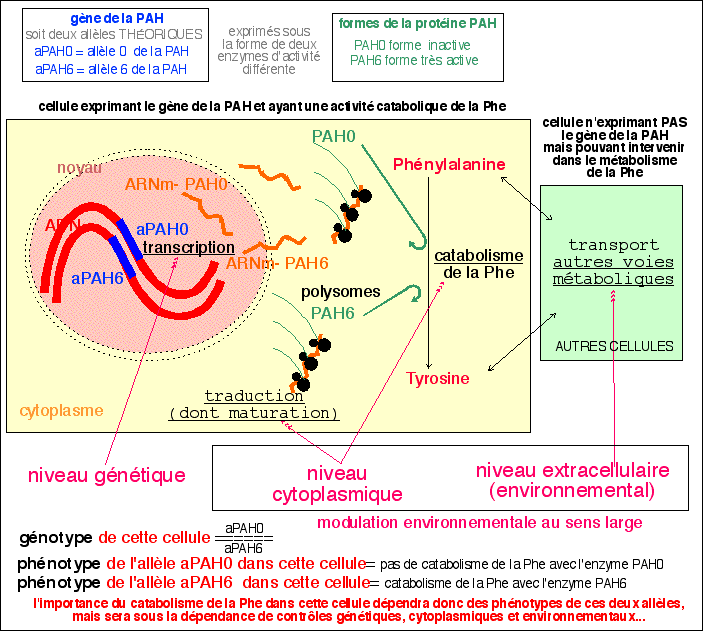
Ces quelques précisions afin de montrer le sens du vocabulaire
employé dans cette page: les niveaux
génétique, cytoplasmique et
environnemental sont définis EN
RÉFÉRENCE À UNE CELLULE DONT ON ÉTUDIE LE
GÉNOTYPE. Sinon, on a vite fait de tout réduire
à un contrôle génétique.
Tout ce qui n'intervient pas directement au niveau des gènes
d'une cellule dont le gène de la PAH est exprimé peut
être qualifié d'environnemental (au sens
le plus souvent employé dans les manuels scolaires)
même si la synthèse de substances qui seraient
impliquées dans ce contrôle nécessite
l'expression d'une autre information génétique (de
cette cellule ou d'une autre cellule). De la même
manière il est judicieux de parler du phénotype d'un
allèle mais fort imprécis de parler du
phénotype d'une cellule car on ne sait pas, sauf
expérience particulière, et pour des conditions de
culture (in vitro) ou d'environnement (in vivo) bien
spécifiques, ce qu'il résultera des informations
génétiques, cytoplasmiques et environnementales.
Supposer par exemple que la cellule synthétise autant de PAH0
que de PAH6 et que le catabolisme qui en résulte soit
moitié moindre que si cette cellule avait deux allèles
aPAH6, est une vue théorique. Parler de phénotype d'un
individu est encore plus mal venu.
Pour situer les réactions métaboliques, voici
un aperçu (d'après Principes de
biochimie, 1994)
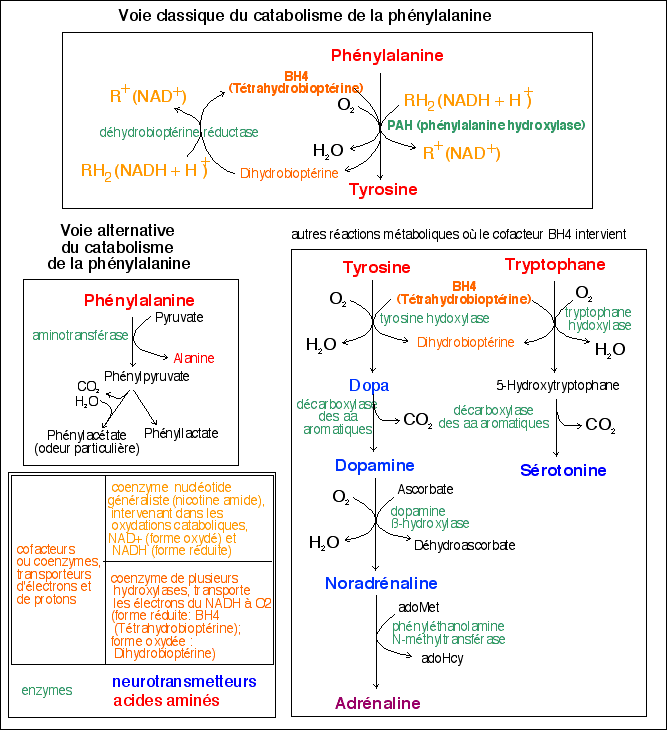
La phénylalanine (Phe) et la tyrosine (Tyr) sont deux
acides aminés nécessaires à l'organisme. Comme
la tyrosine, chez l'homme, est habituellement
synthétisée à partir de la phénylalanine,
seule la phénlylalanine est considérée comme un
aa indispensable, c'est-à-dire que l'homme doit se procurer
dans son alimentation. Les régimes pauvres en Phe suivis par
certains malades contiennent cependant suffisamment de Phe pour
assurer les besoins de synthèse de la Tyr si la PAH n'est pas
déficiente. Sinon il faut ajouter de la Tyr aux
régimes.