|
retour plan cours, accueil, panorama |
|
ou fibrose kystique (CF = cystic fibrosis - OMIM 219700) |
|
en jachère définitive 12/2012
Autres
pages sur ces
questions : |
|||||||||||||||||||
|
Plan |
|
|
|||||||||||||||||||||
|
Le but de cette page n'est en rien de se substituer aux innombrables sites, notamment médicaux, qui donnent des informations extrêmement précises sur les découvertes et espoirs liés à cette maladie. Pour nous, en classe de 1èreS, et dans la continuité du cours de biologie moléculaire, le but est de présenter puis de questionner le modèle de maladie génique. |
|
||||||||||||||||||||||
|
Sources |
|
La
mucoviscidose,
M. Welsh et A.
Smith, 1996
Pour la
science,
février
1996, 220 Sources
d'images et de
données
numériques: |
|||||||||||||||||||||
|
Résumé: En
fait la
mucoviscidose
pose deux
questions
principales : |
|||||||||||||||||||||||
|
1 - La plus fréquente des maladies rares européennes |
|
source
Fanen, 2001 |
|||||||||||||||||||||
|
Histoire
|
|
La description de la mucoviscidose comme une maladie répertoriée, associée à un groupe de symptômes, date de la fin des années 30, 1938 pour une description complète par Andersen. Le nom de fibrose kystique (en anglais cystic fibrosis = CF) qui se réfère à l'association entre une fibrose kystique congénitale du pancréas (anomalies sécrétoires) et des bronchectasies (dilatations des bronches) a été préféré à mucoviscidose, qui se réfère aux sécrétions excessives de mucus épais ("visqueux") au niveau des poumons et de l'appareil digestif notamment.. |
|
En 1953, Di Sant'Agnese met en évidence un excès de chlorure de sodium dans la sueur des enfants atteints. Cette découverte conduira, peu après, à la mise au point du "test de la sueur", principal test de diagnostic positif de la maladie, assez efficace sur les enfants, mais moins sur les nouveau-nés. |
|
Dans les années 80, l'anomalie du transport de sels fut précisée (Quinton 1983) : défaut de perméabilité aux ions chlorure (Cl-) affectant les cellules épithéliales des glandes sudoripares, et au niveau de l'épithélium respiratoire (Knowles 1983) . |
|||||||||||||||||
|
Épidémiologie on
peut suivre
l'épidémiologie
en France sur
le site
de l'INVS ancienne page statistiques/probabilités |
|
La mucoviscidose est la plus fréquente des maladies héréditaires graves de type autosomique récessif (voir ci-dessous)) dans les populations d'origine européenne. Mais il existe de grandes disparités entre pays et entre régions : entre 1/2000 et 1/7000 naissances (29.095 malades en Europe en 2010 - dont 1128 décédés dans l'année - EurocareCF, Journal of Cystic Fibrosis 9 (2010) S5-S21)). En France,on a environ 250 nouveaux cas par an avec un chiffre moyen de 1 malade sur 2500 naissances, ce qui est élevé par rapport à d'autres pays. On évalue le nombre de sujets atteints, quelque soit leur âge, à 4533 personnes en France en 2010, ce qui, sur une population de 64,3 millions, montre bien que cette maladie reste une maladie rare (la prévalence en Europe est évaluée à 0,737/10.000 (voisine de celle des États-Unis qui est de 0,797/10,000) à comparer avec la prévalence totale des cancers estimée à près de 3,4% (340/10.000) de la population française). |
|||||||||||||||||||||
|
|
|
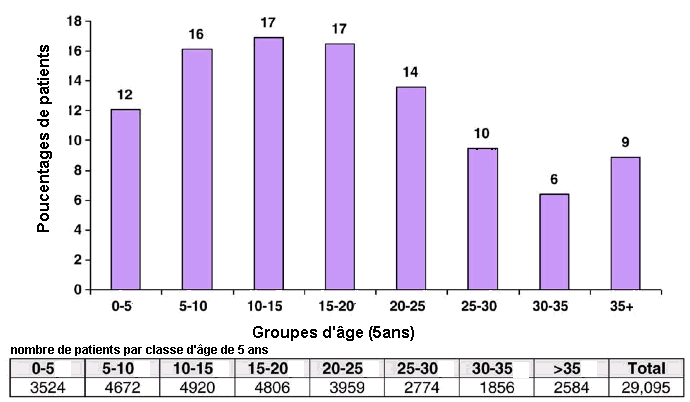
La mucoviscidose est une maladie de l'enfance : malgré l'augmentation de l'espérance de vie, la pyramide des âges des malades en 2010 en Europe montre bien que les jeunes malades sont de loin les plus nombreux (médiane à 20 ans). (source EurocareCF 2010 - image modifiée) |
|||||||||||||||||||||
|
Clinique
|
|
Symptômes et diagnostic C'est une maladie très polymorphe dont les symptômes peuvent être très légers ou très graves et conduire à un mort rapide. Cependant la maladie reste mortelle même si l'espérance de vie n'a cessé d'augmenter . La survie moyenne des patients atteints de mucoviscidose en traitement moderne est actuellement de 40 ans. Avec la transplantation pulmonaire et sa survie moyenne à cinq ans de plus de 70 %, l'âge de ces patients va continuer d'augmenter. Cette maladie héréditaire, à évolution généralement fatale dans l'enfance il y a 20 ans (sans traitement, l'espérance de vie a pour médiane 3-5 ans), est donc devenue une maladie chronique de l'adulte. Causes du décès associées directement à la maladie: affection respiratoire ; infection, grippe, mycose....(70%), pathologie cardiaque (13%), complications de soins médicaux et chirurgicaux (8%), appareil digestif (7%) ... |
|
Chez
10 % des
nouveau-nés
atteints, on
observe un iléus
méconial
(occlusion
intestinale
due à
un méconium
- les
premières
selles de
couleur verte
-
anormalement
épais). 98,% des hommes atteints sont stériles (les canaux déférents sont obstrués et les spermatozoïdes ne passent plus) alors que 80,% des femmes atteintes sont fertiles. Le foie est aussi atteint dans de nombreux cas. |
|
Le
diagnostic
principal
où l'on
mesure un
paramètre
(qualifié
pour cela de
positif en
liaison avec
la philosophie
positive) est
le test
de la sueur.
Ce test repose
sur un
excès
de chlorure de
sodium dans la
sueur du fait
du mauvais
fonctionnement
de
l'épithélium
de la peau qui
peut conduire
à une
forte
déshydratation
en cas
d'exposition
à la
chaleur. Chez
le nourrisson
de moins de 2
mois, le
risque
d'erreur peut
atteindre 30 %
en raison de
la faible
quantité
de sueur
récupérable,
et en
considérant
qu'un test
positif est
efficace
à 100%.
On
considère
qu'un taux de
chlorures dans
la sueur
inférieur
à 40
mmoles/l est
normal. Pour
des taux
compris entre
40 et 60
mmoles/l,
l'interprétation
est douteuse
et il faut
recommencer le
test. Le
diagnostic est
considéré
comme positif
lorsqu'on
obtient des
taux
supérieurs
à 60
mmoles/l (sur
plusieurs
examens
successifs). |
|||||||||||||||||
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
Soins S'il n'existe aucun traitement pour guérir de la mucoviscidose, les soins augmentent grandement l'espérance de vie. Bien évidemment la recherche tente de mettre au point des thérapies plus efficaces pour soigner non plus les symptômes, mais les causes -réelles ou supposées - de la maladie. |
|
Quelques traitements symptomatiques : kinésithérapie respiratoire, antibiothérapie, nébulisation de bronchodilatateurs et mucolytiques, administration d'inhibiteurs de protéases pour les manifestations pulmonaires et apport d'enzymes pancréatiques de substitution et de vitamines pour pallier à l'insuffisance pancréatique. Le recours à une transplantation coeur-poumon voire triple coeur-poumons-foie n'a eu lieu que dans les atteintes très évoluées, mais les résultats sont encourageants, le problème majeur restant le nombre insuffisant d'organes disponibles. |
|
Les thérapies envisagées semblent toutes se faire dans le cadre de l'hypothèse de la maladie génique associée au dysfonctionnement du canal chlore CFTR, ce qui permet d'envisager une thérapie génique et des médicaments ciblés sur l'expression du gène... |
|||||||||||||||||
|
2 - Une maladie héréditaire récessive autosomale au sens de Mendel |
|||||||||||||||||||||||
|
Étant donné le caractère fortement héréditaire de la maladie (le risque d'avoir un enfant atteint dans une famille où l'on a déjà eu un enfant atteint est plus élevé que si aucun malade n'est connu dans la famille), on a très tôt appliqué le raisonnement issu de la théorie chromosomique de Mendel-Morgan datant des années 1900 (voir ancien cours de spécialité TS : page sur Mendel, page sur Morgan). |
|
Selon
cette
théorie
(modèle): |
|
Les génotypes possibles sont donc m+//m+, sain ; m+//m-, porteur, mais de phénotype sain, car l'allèle m- est récessif par rapport à l'allèle m+; m-//m-, la personne est malade. 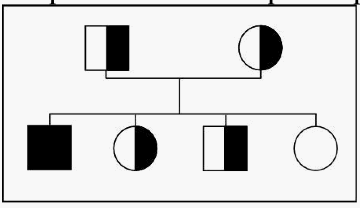 un arbre généalogique très théorique (présentant les 4 cas possibles de descendants) d'une transmission héréditaire d'un gène autosomal récessif (génotypes : porteur sain = figuré semi-noirci; malade = noir, sain = blanc) |
|
Ce formalisme date donc des tout débuts du XXème siècle. C'est le formalisme des maladies héréditaires au sens de Mendel-Morgan, que l'on peut aussi qualifier de maladie génétique, mais en faisant bien attention au sens du mot gène, qui désigne ici un gène héréditaire, portion de chromosome qui porte la maladie.. |
|||||||||||||||||
|
3 - La découverte du gène CFR et le modèle de maladie génique |
|
|
|||||||||||||||||||||
|
|
A la suite des découvertes de la biologie moléculaire et des fonctions de l'ADN (dans les années 1960), la théorie de l'information génétique (voir cours de 1èresS) a développé une autre notion du gène - le gène moléculaire - qui est un portion d'ADN (et non plus de chromosome, du moins dans un premier temps car il y a un changement d'échelle phénoménal) qui est associé (avec certaines séquences de régulation) à la synthèse d'un ARN (par transcription) et le plus souvent d'une protéine (par traduction de l'ARNm transcrit et épissé). C'est donc une unité de synthèse d'un produit (ARN puis protéine). |
|
Depuis 50 ans les biologistes moléculaires tentent de fusionner les deux notions - gène héréditaire / gène moléculaire - sans franchement y arriver, sauf dans quelques cas considérés comme emblématiques, dont la mucoviscidose, du moins en première analyse. |
|
C'est pourquoi la mucoviscidose est un exemple si souvent utilisé : il est considéré comme l'exemple type d'une maladie génique , c'est-à-dire une maladie associée à un seul et unique gène moléculaire. |
||||||||||||||||||
|
|
|
|
|
|
|||||||||||||||||||
|
*Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mullingan RC et al. : Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991;253:202-4 |
|
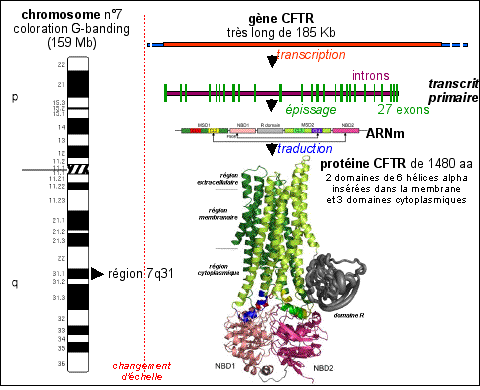
Il faudra attendre 1985 pour que l'on positionne le locus du gène héréditaire (noté CF pour cystic fibrosis) sur le bras long du chromosome 7 grâce à la technique du clonage positionnel (la transmission héréditaire du locus CF est liée à celle d'un site qu'on est arrivé à localiser). En 1989, on localise un gène moléculaire (nommé CFTR : cystic fibrosis transmembrane conductance regulator : régulateur de conductance transmembranaire de la fibrose kystique) dans cette même région de l'ADN du chromosome 7 (bande 7q31) qui code pour une protéine dont on ne connaît pas encore le rôle, mais qui semble associée à la mucoviscidose. C'est en 1991* qu'Anderson et son équipe démontront le rôle de canal chlore pour la protéine CFTR. |
|
|
|||||||||||||||||||
|
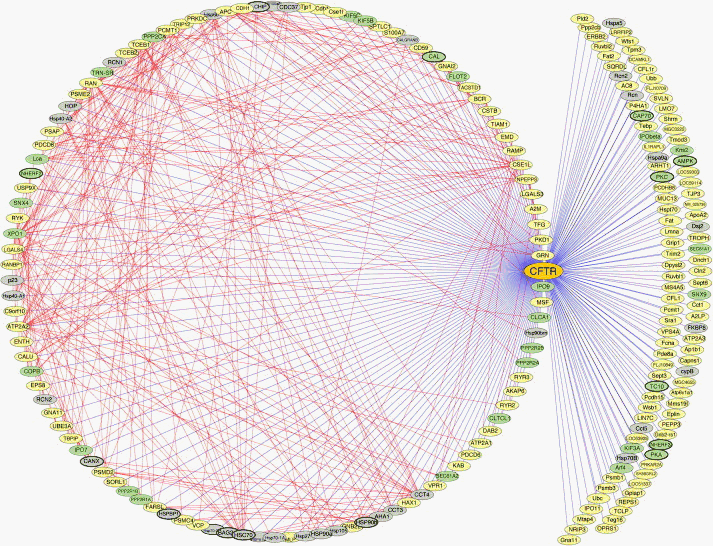
On trouve la CFTR exprimée au niveau de la membrane de nombreuses cellules épithéliales et sanguines. Elle est associée à un réseau de protéines membranaires et cytoplasmiques extrêmement complexe. C'est l'occasion de rappeller ici l'organisation structurée du cytoplasme (voir par exemple ce schéma dans Au-delà de la cellule). La CFTR est en position membranaire et fait partie intégrante d'un COMPLEXE moléculaire dynamique qui comprend aussi le réseau de la cytomatrice (notamment des filaments d'actine). Les produits d'une réaction chimique sont les substrats de la réaction suivante et passent immédiatement d'un complexe enzymatique à l'autre par canalisation (chanelling). |
|
« Bien que la CFTR fonctionne principalement comme un canal à chlore, elle a de nombreux autres rôles notamment en inhibant le transport de sodium par les canaux sodium, en modifiant les canaux à ATP, dans le transport intracellulaire des vésicules, dans l'acidification des organites intracellulaires et en inhibant les canaux à chlore Ca-dépendants CFTR est aussi impliquée dans les échanges bicarbonate-chlorure. Une déficience dans la sécrétion de bicarbonate conduit à la faible solubilité et à l'agrégation des mucus sécrétés dans les lumières des conduits recouverts d'épithélium (pancréas et appareil digestif, bronches et appareil ventilatoire...)» O'Sullivan et Freedman, 2009 (trad. personnelle) - voir ci-dessous des détails sur les hypothèses sur les causes physiologiques de la maladie.
Les fonctions de la protéine CFTR sont loin d'avoir été totalement explorées (voir l'interactome ci-contre ->). Elle semble non seulement avoir un niveau d'expression différent, mais aussi jouer des rôles différents selon le type de tissu épithélial (canaux pancréatiques, tissu bronchique, glandes sudoripares, glandes intestinales, tubules rénaux, appareil génital....) et l'âge (période fœtale, embryonnaire...). |
|
Une
carte
d'activation
des
réactions
chimiques
impliquées
dans de la
fixation de
certaines
espèces
bactériennes
(P.
aeruginosa)
à la
surface
épithéliale
|
|||||||||||||||||||
|
|
|
|
|||||||||||||||||||||
|
polymorphisme génique Pour les biologistes moléculaires qui sont persuadés qu'il y a une identification complète gène héréditaire-gène moléculaire, les allèles sont des séquences du gène. *De même , dans le cadre de la théorie de l'information génétique toute variation de séquence est apellée mutation de façon abusive (voir cours 1èreS) . |
|
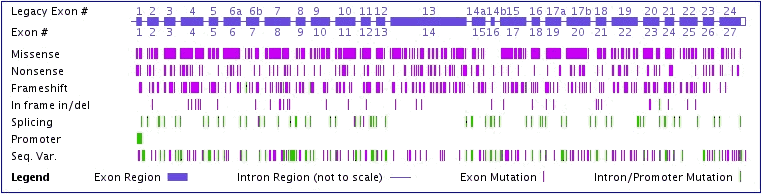
Plus les années passent plus on connaît de mutations* dans le gène CFTR. Pour les 10 mutations les plus fréquentes (dont la principale - la délétion ΔF508 du codon codant pour l'aa n°508 (la phénylalanine) de la CFTR - il existe des kits commerciaux de dépistage rapide. Anticipant sur la liaison génotype-phénotype, que nous traiterons ensuite, les mutations sont classées comme sévères ou légères en fonction de la gravité de la maladie observée. Mais ensuite, il faut bien noter que ce n'est pas la possession de deux "allèles" (forme génique) qui détermine la sévérité de la maladie, c'est juste une appellation. De plus il faut bien considérer que la MAJORITÉ des mutations donnent lieu à des formes fonctionnelles de la protéine CFTR qui ne conduisent donc à aucune déficience métabolique et ne sont donc pas reliées à la maladie (Sequ Var. de la figure ci-dessous). |
|
Comme le veut la théorie d'une maladie héréditaire génique, ceux qui n'ont qu'un seul exemplaire du gène CFTR muté (qu'on qualifiera de "porteur sain") ne sont pas du tout malades (en fait il serait plus exact de dire que la possession d'un seul gène moléculaire fonctionnel de la protéine CFTR empêche toute apparition d'un quelconque symptôme de la maladie). |
|
Selon le formalisme d'une maladie génique la possession de deux allèles récessifs (m-//m-) conduit obligatoirement à une forme de maladie, pus ou moins sévère selon l'environnement et ce que l'on appelle souvent à tort les facteurs épigénétiques qui sont la plupart du temps des facteurs génétiques qui modulent l'activité du gène muté. On fait on obtient un résultat de 87,8 + 11,3 = 99,1 %. Il reste 0,9 % des malades qui n'ont pas mutation détectée. Chez
les malades chez
qui on fait
une analyse
génétique
cherchant
à
déterminer
la
présence
d'une mutation
on obtient : «
Au terme de
cette
recherche, il
reste environ
1 à 2 %
des sujets
atteints de
mucoviscidose
(selon
l'origine
géographique
ou ethnique)
pour lesquels
au moins une
mutation n'est
pas
caractérisée.
Il est
probable qu'il
s'agit chez
ces patients
de mutations
introniques ou
de mutations
situées
dans les
régions
régulatrices
en 5' ou en 3'
du
gène. »
(Ferec, 2011 On voit donc ici que M. Ferec, dans le cadre de l'interprétation génique, suppose que les malades sans mutation sur leur gène CFTR portent des mutations que l'on qualifierait d'épigénétiques. |
|||||||||||||||||
|
|
|
||||||||||||||||||||||
|
Cystic fibrosis, Brian P O'Sullivan, Steven D Freedman Brian O'Sullivan P, Steven Freedman D, 2009, The Lancet, 373, 1891 (les
mutations sont
notées
pour l'ADN
avec le n°
de la base
modifiée,
ancienne et
nouvelle base
séparées
par une
flèche
ou code (X =
del, ins =
insertion); le
signe +
indique le
début
de l'intron
(et le
deuxième
n° la
position de la
base dans
l'intron).
Pour les
chaînes
polypeptidiques
c'est le
n° de l'aa
encadré
par l'ancien
et le nouvel
aa |
|
Si l'on s'efforce de classer les mutations en fonction à la fois de la gravité des symptômes associés et des mécanismes supposés où la mutation provoque une modification physiologique on obtient 5 classes:
|
|
classe
I : la
protéine
n'est pas
synthétisée
; à
cause par
exemple d'un
codon stop qui
bloque la
transcription
(ex: Gly542X)
ou un
défaut
d'épissage
(711+1G
->T) Il faut à nouveau préciser que le nombre de mutations associés à des dysfonctionnements cellulaires est extrêmement réduit (une cinquantaine) par rapport aux 1932 mutations connues (voir figure et site ci-dessus). |
|||||||||||||||||||
|
|
|
« Contrairement à ce que laissent entendre les expressions courantes, et pas seulement sous les plumes incompétentes ou paresseuses, de « gène du cancer », « gène de la mucoviscidose », « gène de l'obésité », etc., les gènes codent pour des protéines - et sûrement pas pour des entités dont la caractérisation selon une causalité linéaire en termes de macromolécules biologiques n'est pas encore connue! En effet, le fait d'établir une corrélation entre deux éléments (le gène et le caractère phénotypique) ne démontre pas par lui-même le lien de causalité qui les unit. L'expression des gènes est donc le mécanisme par lequel un gène (i.e. une séquence de nucléotides) produit une protéine, via une série d'étapes compliquées dont nous ne donnerons que les moments les plus importants.» Marc Silberstein, Jean-Jacques Kupiec et Olivier Gandrillon, De la nécessité du hasard en biologie, in Le Hasard au cœur de la cellule, Editions matériologiques, février 2011 (je suis partisan d'aider à une diffusion aux étudiants et enseignants d'une version allégée (12,2 Mo tout de même) : sitepst(arobase)aliceadsl.fr). |
|||||||||||||||||||||
|
liaison gène moléculaire - symptômes qualifiée de façon erronée de liaison génotype -phénotype (ces deux notions faisant référence à un gène héréditaire et non pas moléculaire)
*
Les partisans
d'une vision
génétique
pourront
notamment
s'enthousiasmer
des millions
dépensés
actuellement
dans le projet
ENCODE
sur le
séquençage
du
génome
humain non
codant. On a
ainsi mis en
évidence
d'innombrables
séquences
d'ADN non
codant
où se
fixent des
protéines
régulatrices
de la
transcription
de
gènes
impliqués
dans de
nombreuses
maladies :
Maurano, et
al., 2012.
Systematic
localization
of common
disease-associated
with variation
in regulatory
DNA. Science
337: 1190-1195
|
|
Les médecins affirmeront sans doute en aparté que les gènes n'expliquent pas tout (on trouve l'affirmation dans quelques présentations de cours de médecine) ou plutôt que tous les symptômes ne sont pas expliqués par le gène de la CFTR. Et pourtant, il est vraiment rare de voir cette question de la liaison génotype-phénotype traitée ou même soulevée. Plus encore, poussés dans leur retranchement, ces mêmes médecins finiront probablement par se replier sur une explication moléculaire génétique du type de celle invoquée par M. Ferec ci-contre : l'épigénétique (qui est alors de la génétique déguisée, notamment par le recours aux "gènes modificateurs") et à l'environnement*. |
|
«
Ces
données
sont
établies
à
partir de
larges
cohortes de
patients
(généralement
issues des
registres de
mucoviscidose),
si elles sont
parfaitement
vérifiées
statistiquement,
il
n'en demeure
pas moins que
l'on peut
observer une
grande
variabilité
à
l'échelle
individuelle.
Ainsi, un
patient
donné
porteur de
deux mutations
sévères
peut
parfaitement
présenter
un
phénotype
relativement
« peu
sévère
»
pendant de
nombreuses
années
et, à
l'inverse, la
présence
d'une mutation
qualifiée
a priori de
« peu
sévère
» chez
un patient
peut parfois
s'accompagner
d'une
évolution
rapide vers
l'insuffisance
respiratoire
terminale. Ces
données
de
corrélation
génotype/phénotype
doivent
être
maniées
avec prudence,
car, si elles
sont
globalement
vraies lorsque
l'on analyse
un groupe de
patients, elles
doivent
être
utilisées
avec la plus
grande
précaution
à
l'échelle
individuelle. |
|||||||||||||||||||
|
|
|
Plus grave encore, dans l'approche soutenue notamment par C. Ferec (voir la 1ère partie de la thèse d'Alix de Becdelievre de 2011 : Contribution à l'amélioration des connaissances sur la relation génotype-phénotype dans la mucoviscidose et caractérisation phénotypique de l'inflammation pulmonaire) on tente de séparer la mucoviscidose, maladie génique qui correspond aux formes plus ou moins sévères de la maladie , avec des pathologies mineures liées à des anomalies du gène CFTR (appelées CFTR-RD : RD signifiant related disorder) conduisant à des formes atypiques ou monosymptomatiques). Cette voie me semble particulièrement pernicieuse puisqu'elle contourne le problème sans questionner le modèle moléculaire de maladie génique. En fait elle renforce la modèle en excluant de l'analyse génétique héréditaire les formes qui ne cadrent pas avec l'idée d'une maladie génique autosomale. La véritable question de la liaison entre le génotype et le phénotype est réellement celle d'un déterminisme qui est loin d'être démontrée : la possession d'un couple de formes gèniques (allèles) dans une cellule à un moment donné peut-elle induire, et avec quelle probabilité, telle caractéristique phénotypique, ici une maladie dans toute sa complexité et sa variabilité. Il ne s'agit pas de nier le rôle des gènes mais de questionner le déterminisme (voir cours de 1èreS). |
|||||||||||||||||||||
|
|
|
- il existe des malades qui n'ont pas de mutation de leur gène CFTR et qui présentent un ensemble de symptômes voisins de ceux qui semblent causés par la perte de fonction de la CFTR; il est sans aucun doute raisonnable de penser que le dysfonctionnement est peut-être situé au même niveau (de la CFTR), même si cela n'est pas prouvé et même s'il ne faut pas se fermer à d'autres interprétations qui ne mettraient pas en jeu directement un canal chlore. |
|
- il existe une grande variabilité dans les symptômes qui n'est pas absolument reliée aux types de mutations portées. Il existe des cas, en plus ou moins grand nombre, de porteurs de double mutation (homozygote) "sévère" qui présentent de formes mineures de la maladie, voire même aucun symptôme, bien que ces cas soient effectivement rarissimes, car on ne va pas chercher les allèles morbides chez les individus sains (sauf lorsque l'on fait des analyses pour des arbres généalogiques en vue d'un conseil génétique). |
|
- la majorité des mutations du gène de la CFTR n'ont pas de relation avec la fonction de la protéine, soit que l'on suspecte qu'elles font partie du polymorphisme, sans empêcher une fonction normale, soit que l'on pense ne pas encore connaître les conséquences de leur présence.. |
|||||||||||||||||
|
|
|
|
|||||||||||||||||||||
|
Conséquences du choix du modèle en terme de diagnostic, de conseil génétique et de recherche |
|
Le conseil génétique est une branche de l'activité médicale qui repose sur le modèle de transmission héréditaire de type Mendel-Morgan mais qui s'est enrichi de la détection des séquences mutées du gène de la CFTR. En reprenant la même notation (m+ =allèle sain ; m- = allèle morbide) on suppose qu'il existe une liste de séquences de la CFTR qui correspondent à un allèle (ms-) qui, à l'état homozygote, causera une forme sévère de la maladie et à l'état hétérozygote avec un autre allèle morbide, de forme modérée (mm-) , causera une forme modérée de la maladie. Je précise à nouveau que ces appellations de "sévère" et "modérée" ne procédent en aucun cas d'une observation directe de l'action sur des cellules en culture par exemple, mais bien de la fréquence de mutations associées statistiquement à des malades atteints de formes sévères ou modérées. C'est en quelque sorte un raisonnement circulaire (qui boucle sur lui-même), particulièrement difficile à débusquer. |
|
La probabilité qu'un individu quelconque soit porteur (m-//m+) est de 1/30 à 1/35 selon les régions. |
|
On fait donc des tableaux de croisement (dits tableaux de gamètes qui indiquent les pourcentages des allèles portés par chaque cellule sexuelle et par chaque cellule œuf résultante:
probabilité
phénotype
sain
= 292/302
+ 2 x 29/302
= 0,9989 |
|||||||||||||||||
|
|
|
|
|
|
|||||||||||||||||||
|
un arbre généalogique très théorique (présentant les 4 cas possibles de descendants) d'une transmission héréditaire d'un gène autosomal récessif (génotypes : porteur sain = figuré semi-noirci; malade = noir, sain = blanc) |
|
Comme il est fréquent que le test génétique soit réalisé pour un enfant en gestation, après l'arrivée d'un enfant malade, les probabilités sont changées: chaque parent étant "porteur sain" (m+//m-). La probabilité d'avoir un enfant atteint devient 1/4.
probabilité
phénotype
sain
= 3 x 1/4 =
0,75 |
|
La détection des mutations chez le fœtus conduit à de très nombreux avortements appelé de façon éhontée "médicaux" qui sont loin de correspondre à un motif médical. |
|
« ...la très grande majorité (96 %) des couples à risque de 1/4; pour lesquels le diagnostic de mucoviscidose fœtale est porté opte pour une demande d'Interruption pour motif Médical de la Grossesse (IMG). » L'usage du terme "mucoviscidose fœtale" est abusif. En effet, on ne sait pas quel sera le diagnostic chez l'enfant, puis l'adulte : il peut être sévère (même pour une mutation rare, voir même en l'absence de mutation) ou modéré (même pour une double mutation grave comme Δ508). C'est tout le problème de la relation génotype-phénotype. |
|||||||||||||||||
|
Comme pour la trisomie 21 où le phénomène est patent, et dans une bien moindre mesure ici, on a tendance à abandonner la recherche de traitement au profit d'une sélection eugénique. |
|
Soit on traite les causes (médecine préventive), soit les symptômes (médecine clinique). Comme on semble avoir renoncé à chercher d'autres causes que génétiques, on peut parfois se contenter du diagnostic génétique (et d'avortement dans 96 % des cas de détection chez le fœtus). Même dans le cas où il n'existe pas de cas dans la famille, mais que l'échographie a permis de poser un diagnostic (intestin hyperéchogène) de suspicion de pathologie, on a des taux très faibles d'association avec une mutation du gène CFTR (dans 3 à 5 % des cas pour ce symptôme...), qui elle non plus d'ailleurs, encore une fois, n'est pas un diagnostic de maladie (je n'ai pas trouvé le chiffre des cas répertoriés d'association avec la maladie CF). |
|||||||||||||||||||||
|
4- Dépasser le modèle génique ? |
|
|
|||||||||||||||||||||
|
|
|
Tant
que l'on
recherchait un
gène
(ou plusieurs)
de ce que l'on
pensait
déjà
être une
maladie
héréditaire
génique
(depuis le
début
du
siècle),
on
était
très
ouvert
à
l'intervention
de plusieurs
gènes
et à
d'autres
interprétations
non
moléculaires
de la maladie. |
|||||||||||||||||||||
|
Les hypothèses physiologiques et cellulaires à l'origine des pathologies de la CF sont habituellement reliées aux différents rôles de la CFTR dont la liste s'allonge, mais si l'on supprime le parti-pris du fonctionnement de la protéine canal, il n'en reste pas moins que ces hypothèses peuvent servir à préciser les causes de la maladie à un autre niveau qu'au niveau moléculaire. |
|
Quatre hypothèses pouvant intervenir ensemble (in O'Sullivan and Freedman, 2009, traduction personnelle): « - l'hypothèse du faible volume d'eau : la réabsorption de l'eau (donc un excès d'hydratation cellulaire) et l'excès de sodium extracellulaire conduiraient à la déshydratation des voies respiratoires.La diminution simultanée de la sortie d'ion chlorure empêche l'épithélium de corriger le volume d'eau au niveau des voies respiratoires. La diminution du volume d'eau entourant la zone ciliée a pour conséquence la réduction de la lubrification de la couche située entre les cellules épithéliales et le mucus, avec une compression des cils par le mucus qui inhibe la formation normale des cils et le renouvellement du mucus par la toux. Selon cette hypothèse le mucus forme des plaques sur l'épithélium qui sont des niches anoxiques favorables à l'hébergement de bactéries, tout particulièrement Pseudomonas aeruginosa. - l'hypothèse de la forte teneur en sel : des ions chlorures et sodium se trouveraient retenus en excès dans le liquide des voies respiratoires. L'augmentation de la concentration en chlorure au niveau périciliaire perturberait la fonction antibiotique naturelle de molécules (comme la ß-défensine1) , permettant ainsi à des bactéries qui sont normalement détruites dans les voies respiratoires de persister dans les poumons. |
|
- l'hypothèse de la dérégulation des la réponse immunitaire de l'hôte : en effet des concentrations anormalement élevées de médiateurs de l'inflammation sont observés dans les cultures de cellules atteintes de CF pourtant non infectées. De plus, des études de lavage de poumons ont montré que l'inflammation présente chez les enfants de moins de 4 semaines est indépendante d'une quelconque infection. Une augmentation des molécules favorisant l'inflammation, comme les interleukines 8 et 6, le facteur de nécrose des tumeurs alpha, et des métabolites de l'acide archidonique, a été observée chez des patients atteints de CF. La stimulation de la voie du facteur nucléaire kB, une hyperréactivité des plaquettes et des anomalies dans l'apoptose des neutrophiles, ont été signalées. En même temps la concentration de substances bnaturelles anti-inflammatoires comme l'interleukine 10, la lipoxine et l'acide docosahexaenoique, est réduite, conduisant à un déséquilibre entre les médiateurs pro et anti-inflammatoires qui favorise dans tous les cas l'inflammation. |
|
- une dernière hypothèse repose sur un mécanisme de prédisposition à l'infection : si P. aeruginosa se fixe à la protéine fonctionnelle CFTR chez les individus sains, ce qui initie la réponse immunitaire innée, rapide et autocontrôlée, chez les patients atteints de CF la fixation de P. aeruginosa et de Staphylococcus aureus qui augmente au niveau de l'épithélium des voies respiratoires est favorisée par l'augmentation de l'asialo-GM1 et se fait sans activer la réponse immunitaire initiée par la CFTR. Ainsi, lors d'une CF, la réponse rapide et auto-contrôlée qui devrait éliminer P. aeruginosa des voies aériennes est perdue en même temps que l'attachement des bactéries à la surface de l'épithélium est renforcé.» |
|||||||||||||||||
|
en travaux |
|
On voit donc que ces hypothèses, même reliées au fonctionnement de la CFTR (intervenant aussi bien dans l'osmorégulation que comme facteur immunitaire...), montrent que les perspectives sur des voies non moléculaires ne sont pas fermées. Les hypothèses centrées sur les problèmes de la couche limite des cellules épithéliales (zone ciliée et glycocalix...) peuvent être abordées par des voies qui ne sont pas celles de la biologie moléculaire : la vision que l'on a de l'eau extracellulaire, comme un fluide plus ou moins concentré est particulièrement erronée (voir page sur l'eau dans la cellule et page sur l'hydratation). Les hypothèses centrées sur le rôle immunitaire de la CFTR permettent aussi d'envisager des explications qui ne se cantonnent pas au niveau moléculaire, même si l'immunité est parfois submergée par la recherche de marqueurs membranaires et autres cytokines. L'immunité est d'abord un phénomène cellulaire, les molécules n'étant que les outils ou les traces des phénomènes (voir quelques idées dans l'ancienne page sur l'immunité et dans la page sur le VIH). |
|||||||||||||||||||||
|
|
|
Si l'on persiste dans le modèle moléculaire, on arrive -peut-être- à l'idée que les interactions entre gènes sont si nombreuses (voir CFTR interactome) que l'on se demande bien comment une mutation plus ou moins invalidante peut être STABILISÉE chez les malades. C'est encore une fois de la complexité que naît l'incompréhension. S'il n'y a pas un seul gène, mais de nombreux gènes en interaction, comment un gène unique est-il devenu le point central de la recherche. Il n'est pas possible de penser que l'on soit passé à côté d'autres gènes essentiels, car alors, comment expliquer la forte association entre certaines mutations de ce gène et la maladie. Que la corrélation génotype-phénotype soit plus ou moins bonne est très facilement explicable, au moins de façon floue, par le recours à l'extrême complexité de l'expression de l'information génétique. Une fois encore on se demande s'il ne serait pas utile de faire une étude de répartition des allèles dans une population qui n'est pas à risque avec un échantillonnage suffisamment important (et non quelques témoins comme c'est l'usage). |
|
Une idée classique serait que les malades ne sont pas malades parce qu'ils ont tel allèle, mais bien parce qu'ils ne compensent pas le dysfonctionnement. Il devient alors plus facile d'envisager d'autres causes qui ne sont pas directement reliées au canal CFTR. On peut rester dans le modèle moléculaire, que l'on peut aussi modifier à l'aide du modèle stochastique (théorie ESG, de l'expression stochastique des gènes). |
|||||||||||||||||||
|
|
|
|
|
|
|||||||||||||||||||
|
en travaux |
|
Une
dernière
piste : |
|||||||||||||||||||||


{kind=link}