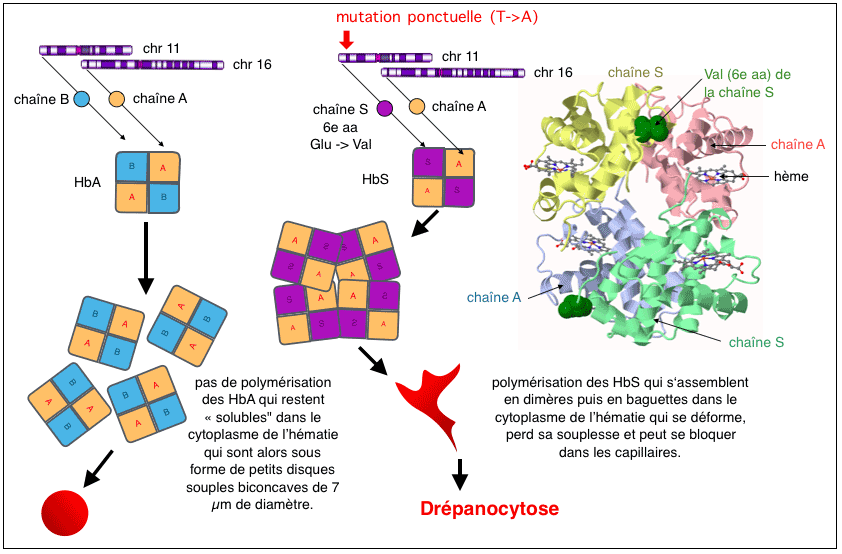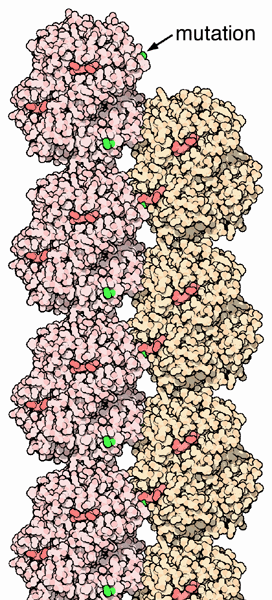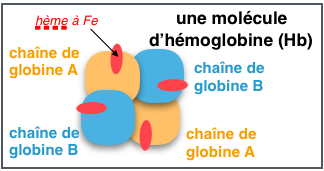
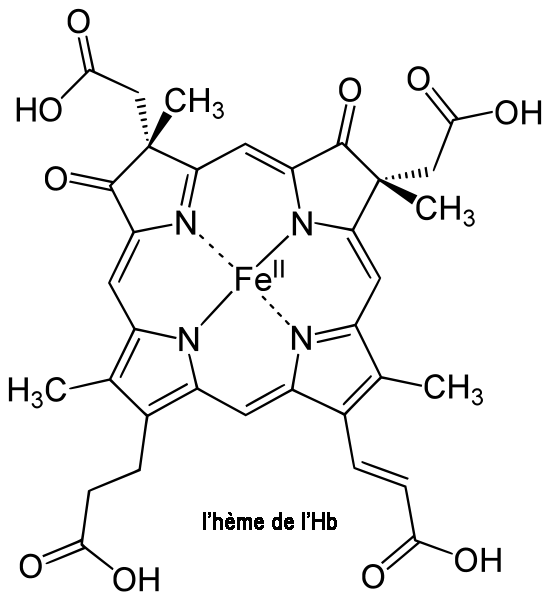
1 - De la séquence des nucléotides de l'ADN à la séquence des acides aminés
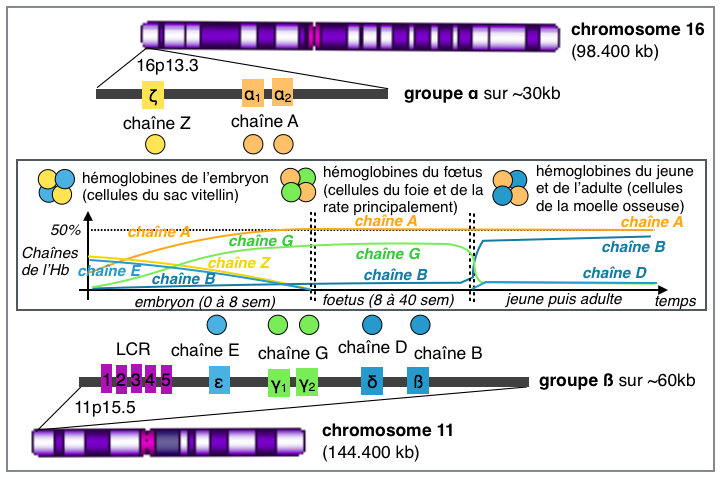
Les
gènes
des
chaînes
de globine
de
l'hémoglobine
sont
regroupés
en deux
groupes (clusters)
positionnés
sur deux
chromosomes
différents
:
- les gènes du groupe α codant pour les chaînes de type A sont sur le chromosome 16,
- alors que ceux du groupe β codant pour les chaînes de type B sont sur le chromosome 11.
- les gènes du groupe α codant pour les chaînes de type A sont sur le chromosome 16,
- alors que ceux du groupe β codant pour les chaînes de type B sont sur le chromosome 11.
Dans l'illustration ci-contre --> j'ai volontairement limité le nombre de gènes (image plus complète). Le groupe LCR dans le groupe β désigne des gènes régulateurs (locus control region).
Au cours de la vie embryonnaire ce sont les chaînes A et Z viennent du groupe α alors que les chaînes E, G et B sont synthétisées à partir du groupe β. Les gènes α1 et α2 sont identiques et donc les chaînes A1 et A2 ont la même séquence.
Chez le fœtus les chaînes A sont dominantes pour le groupe α et les chaînes G pour le groupe β.
À la naissance, laors que les chaînes A restent presque exclusives pour les gènes du groupe α, on observe un basculement pour les chaînes issues des gènes du groupe β : les chaînes G diminuent alors que la chaîne B devient majoritaire (avec un peu de chaîne D).
Il
existe une
très
grande variabilité
individuelle
et
populationnelle
des
différentes
chaînes
de l'Hb,
même
à
l'état
adulte. Chez
un adulte on
trouve
habituellement
:
- 97% de molécules d'HbA1 - AABB notées A2B2 (2 chaînes A et 2 chaînes B)
- 3% de molécules d'HbA2 - AADD notées A2D2 (A2D2= 2 chaînes A avec 2 chaînes D) et plus rarement d'HbF (<1%) AAGG notées A2G2 (A2G2= HbF, hémoglobine fœtale = 2 chaînes A et 2 chaînes G). Il n'y a pas de différence d'efficacité entre ces 3 molécules d'Hb (HbA1, HbA2 et HbF). Si les chaînes A et B diffèrent par plusieurs dizaines d'aa (voir ci-dessous), les chaînes G diffèrent entre elles par un unique acide aminé en position 136, alanine pour G1 et glycine pour G2.
- 97% de molécules d'HbA1 - AABB notées A2B2 (2 chaînes A et 2 chaînes B)
- 3% de molécules d'HbA2 - AADD notées A2D2 (A2D2= 2 chaînes A avec 2 chaînes D) et plus rarement d'HbF (<1%) AAGG notées A2G2 (A2G2= HbF, hémoglobine fœtale = 2 chaînes A et 2 chaînes G). Il n'y a pas de différence d'efficacité entre ces 3 molécules d'Hb (HbA1, HbA2 et HbF). Si les chaînes A et B diffèrent par plusieurs dizaines d'aa (voir ci-dessous), les chaînes G diffèrent entre elles par un unique acide aminé en position 136, alanine pour G1 et glycine pour G2.
Pour les chaînes anormales (hémoglobinoses et thalassémies) et les maladies (hémoglobinopathies), voir la 4e partie de cette page.
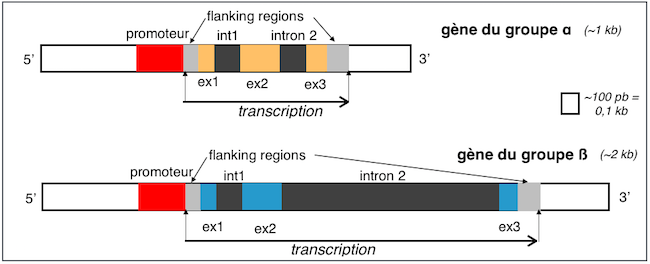
La
structure de
tous les gènes
de globine
est similaire
(<-
image
ci-contre),
trois exons et
deux introns (avec
des
régions
répétées
en 3' et en 5'
= flanking
regions),
la principale
différence
étant que le
deuxième
intron est
beaucoup plus
long que le
premier dans
la famille
ß
(intron 1 =
120-140pb,
intron 2 = 900
pb) alors
qu'ils sont du
même ordre de
grandeur,
moins de 150
pb (paires de
bases), dans
la famille
α. Du
fait de la
présence
de ce long
exon les
gènes
de type β
sont donc deux
fois plus
longs que les
gènes
du groupe
α.
Les transcrits primaires subissent un épissage et l'ajout d'une coiffe et d'une queue puis migrent dans le cytoplasme de l'érythroblaste.
La traduction à lieu au niveau de polysomes libres dans le cytoplasme.
Les chaînes de globine fixent d'abord leur hème avant de s'assembler en tétramère pour former les molécules d'hémoglobine.
Les gènes associés à la synthèse de l'hème sont ceux des enzymes qui interviennent dans sa synthèse; par exemple la ferrochélatase (⎋FECH) qui lie le Fe2+ à la protoporphyrine, elle-même synthétisée en plusieurs étapes à partir d'un produit mitochondrial, le succinyl-CoA, et la glycine, un aa. sous le contrôle de 7 enzymes supplémentaires dont les gènes sont situés sur différents chromosomes (1, 3, 9, 10, 11 et 18).
Les transcrits primaires subissent un épissage et l'ajout d'une coiffe et d'une queue puis migrent dans le cytoplasme de l'érythroblaste.
La traduction à lieu au niveau de polysomes libres dans le cytoplasme.
Les chaînes de globine fixent d'abord leur hème avant de s'assembler en tétramère pour former les molécules d'hémoglobine.
Les gènes associés à la synthèse de l'hème sont ceux des enzymes qui interviennent dans sa synthèse; par exemple la ferrochélatase (⎋FECH) qui lie le Fe2+ à la protoporphyrine, elle-même synthétisée en plusieurs étapes à partir d'un produit mitochondrial, le succinyl-CoA, et la glycine, un aa. sous le contrôle de 7 enzymes supplémentaires dont les gènes sont situés sur différents chromosomes (1, 3, 9, 10, 11 et 18).
La structure
primaire
désigne
la
séquence
ou l'ordre des
aa de chaque
chaîne.
Les
chaînes
de type A ont
141 aa et les
chaînes
de type B 146
aa.
Pour comparer
les
séquences
il faut
utiliser des
logiciels, par
exemple ceux
accessibles en
ligne (comparaison
de 2
séquences
ou plus).
Choisir
d'abord les
séquences
à
comparer (par
exemple ici
en entrant le
code PDB (ou
Uniprot) des
protéines
à
comparer :2hhb
qui est de la
forme HbA1 ou
AABB ou A2B2),
puis copier
les
séquences
dans la page
de calcul
(deux premiers
résultats
de la page).
Ici les aa sont représentés par leur lettre et les alignements sont optimisés par le logiciel : * signifie 2 aa identiques (il est clair qu'il existe d'autres solutions, même si elles sont considérées comme moins pertinente car présentant une différence nécessitant un "chemin" plus long). On voit ainsi que les deux chaînes A et B de cette l'Hb humaine sont très voisines (60 aa alignés identiquement). Ces similitudes sont interprétées en terme d'évolution moléculaire (duplication de gènes et variation allélique).
Tableau comparant les séquences des chaînes A, B, D, G1 et G2
| comparaison des séquences des chaînes | A | B | D | G1 | G2 |
| A | id | 2aa
en plus pour A
et 7(1+6) aa
en plus pour
B, ce qui fait
au total 141
aa pour A et
146 aa pour B; 60 aa identiques en même position soit 43% d'homologie |
2
aa en plus
pour la
chaîne D
et 7 aa en
moins pour la
chaîne A 59 aa identiques/145 soit 41% d'homologie |
2
aa en plus
pour la
chaîne
G1 et 7 aa en
moins pour la
chaîne A 56 aa identiques/145 soit 40% d'homologie |
2
aa en plus
pour la
chaîne
G2 et 7 aa en
moins pour la
chaîne A 56 aa identiques/145 soit 40% d'homologie |
| B |
|
id | 10
aa
différents
sur 146 93% d'homologie |
107 aa identiques /146 soit 73% d'homologie | 108 aa identiques /146 soit 74% d'homologie |
| D |
|
|
id | 41 aa différents sur 146 soit 72% d'homologie | 40 aa différents sur 146 soit 73% d'homologie |
| G1 |
|
|
|
id | identiques sauf 1aa n°136 (G2 possède une Gly à la place de l'Ala pour la chaîne G1) - 99% d'homologie |
| G2 |
|
|
|
|
id |
La composition des Hb synthétisées ne peut pas constituer un phénotype moléculaire.
Cette question est délicate car on a vite fait de confondre les notions héréditaires (liés à l'individu) et celles définies aux niveaux cellulaires et moléculaires.
Dans l'hérédité mendelienne-morganienne (ancien cours spécialité), une gène héréditaire est une particule portée par un chromosome (le chromosome est considéré comme un groupe de liaison de différents caractères portés par des gènes situés sur ce même chromosome). Chaque gène héréditaire gouverne un caractère qui peut se trouver sous plusieurs formes ou allèles. Les rapports entre les allèles sont récessivité ou dominance ou encore codominance.
Dans la théorie de l'information-programme génétique, qui est celle de tous vos manuels scolaires, on tente d'assimiler le gène moléculaire au gène héréditaire et les allèles aux différentes séquences des gènes. Le gène héréditaire devient virtuel, seul reste le gène moléculaire qui est une séquence d'ADN. Mais on sait maintenant qu'un gène héréditaire peut correspondre à tout un réseau de gènes et qu'un allèle, défini au niveau d'un caractère, est déjà un phénotype, qui doit donc reposer de plusieurs gènes moléculaires ayant chacun plusieurs séquences possibles.
Depuis cette confusion de vocabulaire, on est toujours induit en erreur à chaque fois que l'on utilise le mot de gène et d'allèle dans le sens de la théorie de l'information-programme génétique.

Plusieurs gènes doivent être activés pour faire une molécule d'Hb.
Chaque gène de l'Hb ne code que pour une chaîne et, s'il est transcrit et traduit, il donne un polypeptide plus ou moins fonctionnel, mais jamais seul. Il faut deux gènes pour donner une Hb fonctionnelle et donc un caractère phénotypique (sauf cas particuliers de quelques molécules d'Hb ayant 4 chaînes identiques). Il est impossible de définir un phénotype qui ait un sens (sauf classificatoire) au niveau de l'expression d'un gène d'une seule chaîne. La molécule d'Hb est gouvernée par 2 gènes pour les globines et un certain nombre de gènes pour ses hèmes. ON NE DEVRAIT PAS PARLER D'UN GÈNE DE L'HB MAIS BIEN D'UN GÈNE D'UNE CHAÎNE DE GLOBINE.
Le niveau de synthèse de chaque gène de globine ne peut être un allèle.
Si l'on veut fusionner les notions héréditaires et moléculaires il est nécessaire de se limiter au phénotype de la molécule complète d'Hb sans entrer dans le détail des gènes moléculaires qui la contrôlent.
On imagine pouvoir définir facilement le phénotype moléculaire d'une cellule en faisant non pas la liste des chaînes, mais celle des molécules d'Hb qu'elle synthétise à un moment donné au cours de sa vie. Attention, il ne s'agit PAS du phénotype cellulaire qui prend en compte toutes les modifications sur la forme ou la physiologie de cette cellule, selon sa composition moléculaire d'Hb. Mais une difficulté insurmontable intervient : tout individu posséde TOUJOURS DEUX exemplaires de chaque groupe de gènes, en provenance de ses parents. Il n'y a pas d'allèles possible au sens de séquence différente donnant un produit différent (voir la variabilité dans le dernier paragraphe). Un allèle sera donc caractérisé par le niveau d'expression de chaque Hb et ce n'est pas la définition d'un allèle au sens moléculaire de séquence d'un gène.
Si l'on souhaitait utiliser le vocabulaire de la génétique mendelienne-morganienne en parlant de dominance, codominance, dominance incomplète, récessivité... on trouverait toujours les mêmes "allèles" chez tous - au polymorphisme près-, mais plus ou moins exprimés. Comment déterminer l'apport de chacun des parents pour en faire une analyse héréditaire ? Si l'on fait une analyse des Hb présentes dans son sang en prélèvant une petite quantités de sang, on supposera que celui-ci est homogène et représentatif de l'ensemble de l'organisme. Après éclatement des hématies, on sépare les différentes MOLÉCULES par électrophorèse (ce ne sont pas les chaînes mais les molécules qui migrent plus ou moins loin en fonction de leur masse et de leur charge ---> figure ci-contre).
Mais cette technique ne détermine pas quantitativement les différentes Hb. Et pourtant, les allèles ne pourront qu'être définis quantitativement et non pas qualitativement. Je n'ai trouvé aucun document à ce sujet.
Si l'on observe chez un sujet une plus grande quantité d'HbA2 par rapport à d'autres individus, il sera impossible de relier cela à une dominance ou une récessivité. Il y a clairement une impasse dans cette vision de l'hérédité des Hb.
Remarque : si l'on peut dire que, pour une molécule la notion de phénotype moléculaire d'un individu a un certain sens, celui-ci ne peut que présenter qu'un signal moyenné et partiel. Il est clair que le modèle héréditaire mendélien-morganien n'est pas adapté à une compréhension des interactions entre gènes moléculaires. Les véritables questions - pour lesquelles je n'ai pas de réponse - demeurent : quelles cellules expriment quels gènes à quel niveau d'expression et à quel moment, chez un individu donné ? Quelle est la variabilité de l'expression de ces gènes au niveau des populations ? Y-a-t-il une variabilité liée à des paramètres externes (on dirait "épigénétiques", mais je pense à des paramètres physiologiques ou de résistance individuelle...).
Pour dire les choses clairement d'une autre manière : la variabilité de la synthèse des chaînes de globine n'est pas un caractère héréditaire au sens de Mendel-Morgan.
Remarque : la molécule de myoglobine, qui n'a qu'une sous-unité présente les mêmes difficultés que l'Hb en ce qui concerne l'hérédité. Le gène de la myoglobine (MB - référence MYG_HUMAN, P02144 dans la base Uniprot) est situé sur le chromosome 22 (position q12.3) sur le brin inverse. Chez l'homme il donne lieu à 10 transcrits différents avec 2 ou 3 grands introns conduisant à des protéines entre 74 et 154 aa. C'est un gène extrêmement conservé dans les différentes espèces conduisant à des variants utilisés depuis les tous débuts de l'évolution moléculaire pour crére des arbres phylétiques de ressemblance (voir par exemple le site de l'ac-Nice qui utilise Jalview comme comparateur de séquence et pour construire les arbres phylétiques). Ces arbres phylétiques n'ont strictement rien à voir avec la transmission héréditaire des gènes qui est supposée identique au sein des espèces et avec mutation entre espèces différentes. On ignore encore, comme pour l'Hb, l'équipement génétique des différentes cellules de l'individu et l'utilisation qu'elle fait de ses gènes.
Dans la 4e partie on verra d'autres allèles (formes de l'Hb) correspondant à des associations de chaînes rares conduisant parfois à des maladies que l'on a assimilé à des allèles héréditaires par le même artifice.
2 - De la structure primaire à la forme de la protéine
L'hémoglobine
est le type
même de
protéine
complexe avec
4 niveaux de
structure (voir
page
spéciale).
Elle
est
composée
de 4
chaînes
identiques 2
à
2 (2
chaînes
A -
malheureusement
nommées
A et C dans le
fichier PDB
des
molécules
-
et 2
chaînes
B -
malheureusement
nommées
B et D dans ce
même
fichier PDB).
| Quelques
commandes pour
JSmol
Eau : visible,
cachée
Hème
:
compact,
boules
et
bâtons
(couleurs
de
références
page
JSmol)Globine(s) : compact, squelette, boules et bâtons, acides aminés colorés, acides aminés CPK |
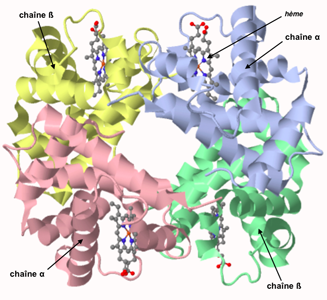 |
- une seule chaîne de globine A humaine [menu "chaînes colorées" ou ici];
- deux chaînes de globine A et B humaines oxygénées associées: une chaîne A (bleue) et une chaîne B (verte) [menu "chaînes colorées" ou ici], formant ainsi une demie molécule d'hémoglobine humaine oxygénée ;
- la molécule complète d'hémoglobine humaine complète désoxygénée : 2 chaînes de type A (A: bleue et C: rose) et 2 chaînes B (B: verte et D:jaune) [menu "chaînes colorées" ou ici].
Remarque: protéine de transport de l'Hb soluble dans le sang : l'haptoglobine (synthétisée par le foie) représentée ici sous forme de 4 molécules d'haptoglobine transportant 4 chaînes A et 4 chaînes B associées en 4 dimères (4f4o.pdb).
Si
la structure
primaire repose
sur la
linéarité
de la
molécule
polypeptidique
qui est avant
tout un copolymère
de 20 types
d'aa (voir
la
liaion
peptidique sur
la page
"peptides"),
3 niveaux de
structure
supplémentaires
expliquent la
forme de la
molécule
d'Hb dans
l'espace :
- Structure secondaire : on notera que les chaînes sont formées chacune de 7 hélice α pour les chaînes A et de 8 hélices α pour les chaînes B, hélices reliées par des zones plus ou moins déroulées et dessinant des coudes.
- Structure tertiaire : l'hème est lié de façon covalente à un aa de type histidine (His) (aa n°87 de la chaîne) au sein d'une poche hydrophobe. La forme de chaque chaîne est stabilisée par des liaisons faibles (on peut voir en utilisant la chaîne de globine A avec le modèle boules et bâtons qu'il n'y a pas de pont disulfure).
- Structure quaternaire : elle résulte de l'association des 4 chaînes de globine par des liaisons faibles. Les chaînes non homologues sont fortement intriquées et liées alors que les chaînes homologues ont peu de contacts entre elles.
Fichier sur la
base
européenne
EMBL-EBI : (2hhb.pdb);
pour
l'organisation
tétramérique
,
l'organisation
dans l'espace
ou topologie
est de type
2/2: 2
sous-unitées
répétées.
Le site http://www.periodicproteincomplexes.org
permet de voir
qu'il existe
389
protéines
de topologie
similaire. - Structure secondaire : on notera que les chaînes sont formées chacune de 7 hélice α pour les chaînes A et de 8 hélices α pour les chaînes B, hélices reliées par des zones plus ou moins déroulées et dessinant des coudes.
- Structure tertiaire : l'hème est lié de façon covalente à un aa de type histidine (His) (aa n°87 de la chaîne) au sein d'une poche hydrophobe. La forme de chaque chaîne est stabilisée par des liaisons faibles (on peut voir en utilisant la chaîne de globine A avec le modèle boules et bâtons qu'il n'y a pas de pont disulfure).
- Structure quaternaire : elle résulte de l'association des 4 chaînes de globine par des liaisons faibles. Les chaînes non homologues sont fortement intriquées et liées alors que les chaînes homologues ont peu de contacts entre elles.
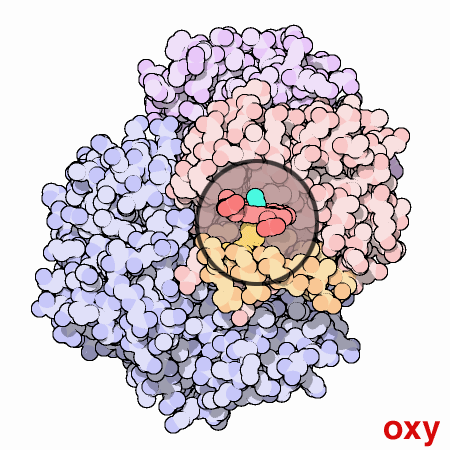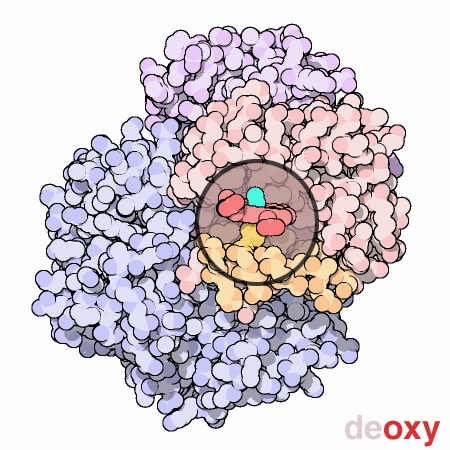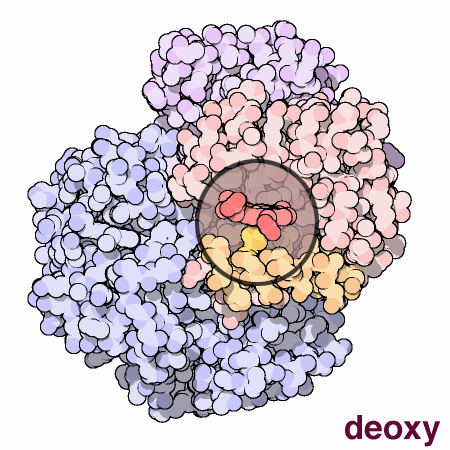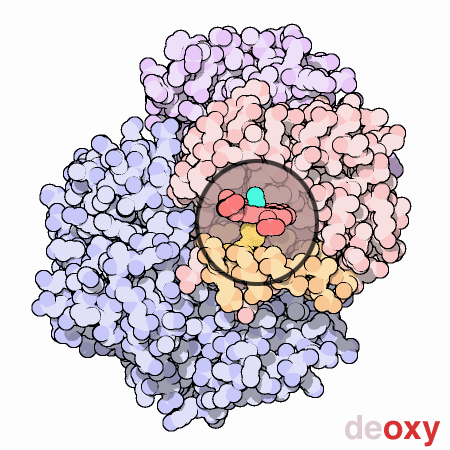
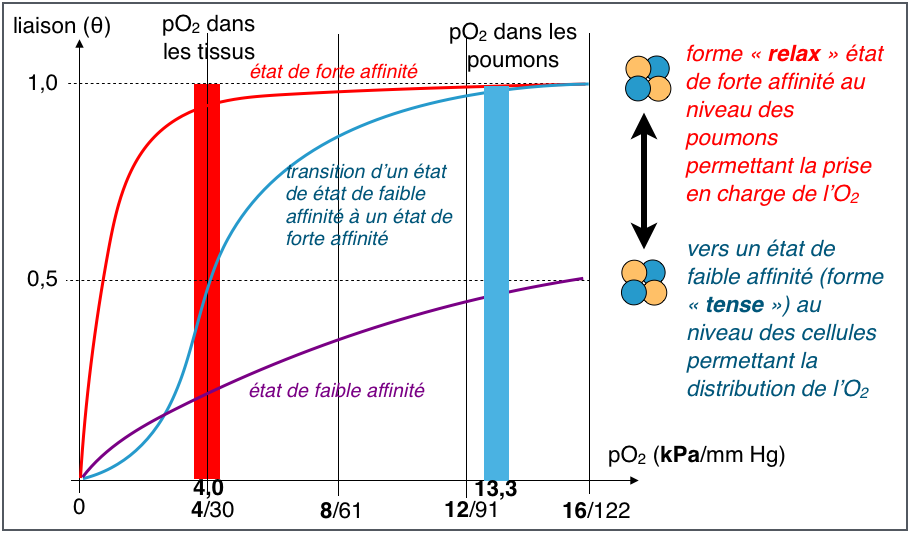
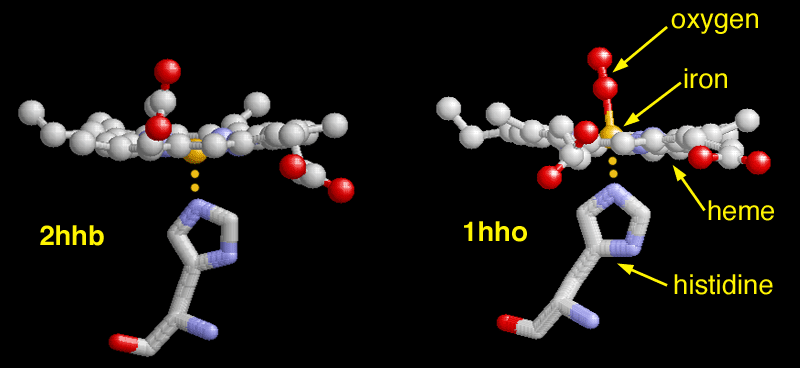 Lorsque
le
dioxygène
se
fixe
sur le FeIII,
l'atome de Fe
est
tiré
vers la
molécule
de
dioxygène
lègèrement
au-dessus du
plan de
l'hème,
ce qui fait
que
l'histidine
à
laquelle le Fe
est lié
par covalence
en-dessous du
plan de
l'hème,
est à
son tour tirée
vers le groupe
Fe-O2,
ce qui
provoque le
mouvement de rotation
de toute la
chaîne.
Ce changement
visible dans
l'image
ci-dessus
Lorsque
le
dioxygène
se
fixe
sur le FeIII,
l'atome de Fe
est
tiré
vers la
molécule
de
dioxygène
lègèrement
au-dessus du
plan de
l'hème,
ce qui fait
que
l'histidine
à
laquelle le Fe
est lié
par covalence
en-dessous du
plan de
l'hème,
est à
son tour tirée
vers le groupe
Fe-O2,
ce qui
provoque le
mouvement de rotation
de toute la
chaîne.
Ce changement
visible dans
l'image
ci-dessus